Institute of Human Genetics, University Hospital Erlangen, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany.
Institute of Human Genetics, University of Leipzig Hospitals and Clinics, Leipzig, Germany.
Eur J Hum Genet. 2022 Dec;30(12):1413-1422. doi: 10.1038/s41431-022-01177-9. Epub 2022 Sep 13.
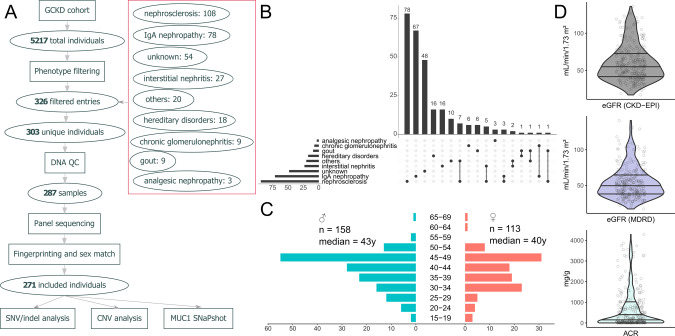
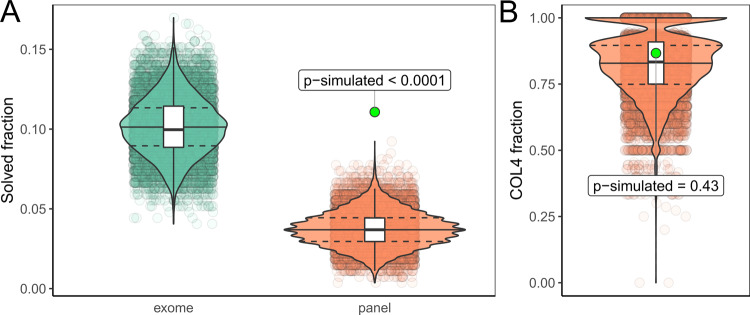
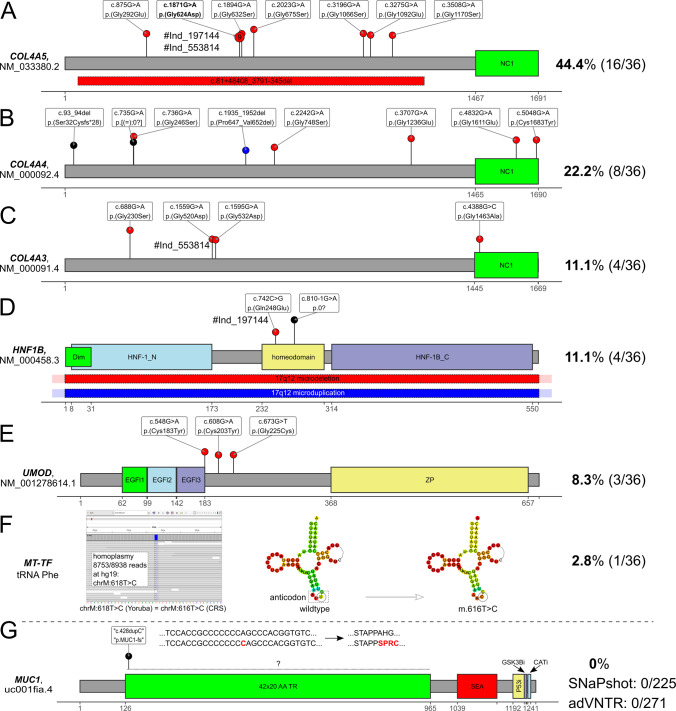
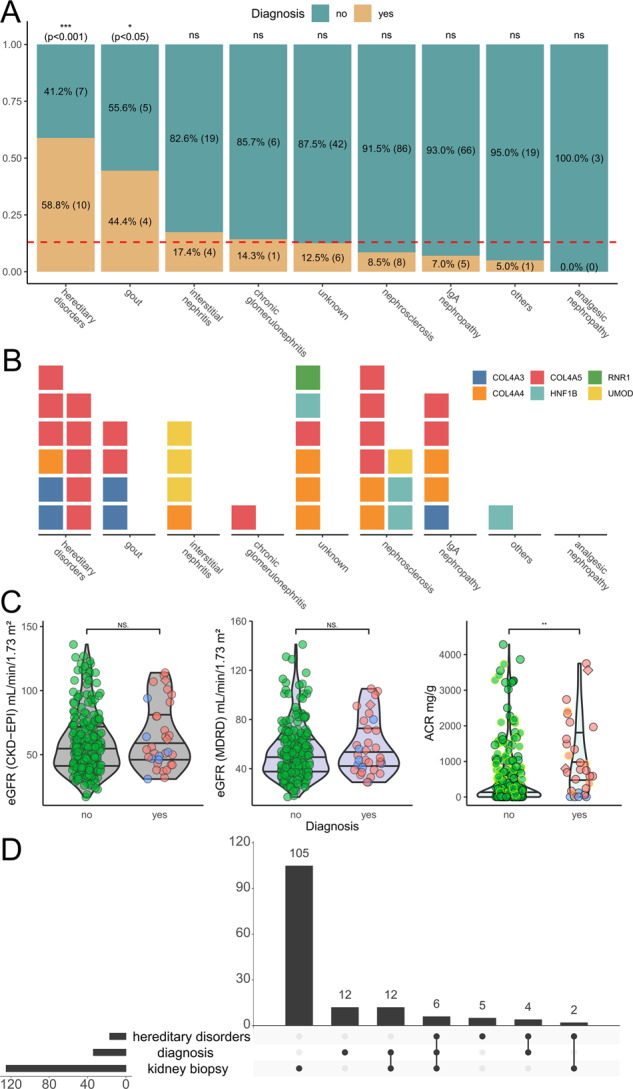
Hereditary chronic kidney disease (CKD) appears to be more frequent than the clinical perception. Exome sequencing (ES) studies in CKD cohorts could identify pathogenic variants in ~10% of individuals. Tubulointerstitial kidney diseases, showing no typical clinical/histologic finding but tubulointerstitial fibrosis, are particularly difficult to diagnose. We used a targeted panel (29 genes) and MUC1-SNaPshot to sequence 271 DNAs, selected in defined disease entities and age cutoffs from 5217 individuals in the German Chronic Kidney Disease cohort. We identified 33 pathogenic variants. Of these 27 (81.8%) were in COL4A3/4/5, the largest group being 15 COL4A5 variants with nine unrelated individuals carrying c.1871G>A, p.(Gly624Asp). We found three cysteine variants in UMOD, a novel missense and a novel splice variant in HNF1B and the homoplastic MTTF variant m.616T>C. Copy-number analysis identified a heterozygous COL4A5 deletion, and a HNF1B duplication/deletion, respectively. Overall, pathogenic variants were present in 12.5% (34/271) and variants of unknown significance in 9.6% (26/271) of selected individuals. Bioinformatic predictions paired with gold standard diagnostics for MUC1 (SNaPshot) could not identify the typical cytosine duplication ("c.428dupC") in any individual, implying that ADTKD-MUC1 is rare. Our study shows that >10% of selected individuals carry disease-causing variants in genes partly associated with tubulointerstitial kidney diseases. COL4A3/4/5 genes constitute the largest fraction, implying they are regularly overlooked using clinical Alport syndrome criteria and displaying the existence of phenocopies. We identified variants easily missed by some ES pipelines. The clinical filtering criteria applied enriched for an underlying genetic disorder.
遗传性慢性肾脏病 (CKD) 的发病率似乎高于临床认知。在 CKD 队列的外显子组测序 (ES) 研究中,大约 10%的个体可以发现致病性变异。表现为无典型临床/组织学表现但存在肾小管间质纤维化的肾小管间质性肾脏疾病尤其难以诊断。我们使用靶向 panel(29 个基因)和 MUC1-SNaPshot 对来自德国慢性肾脏病队列的 5217 名个体中按年龄和疾病实体选择的 271 个 DNA 进行了测序。我们发现了 33 个致病性变异。其中 27 个(81.8%)位于 COL4A3/4/5 基因中,最大的一组是 15 个 COL4A5 变异,其中 9 个无亲缘关系的个体携带 c.1871G>A,p.(Gly624Asp)。我们在 UMOD 中发现了 3 个半胱氨酸变异,在 HNF1B 中发现了一个新的错义变异和一个新的剪接变异,以及同源 MTTF 变异 m.616T>C。拷贝数分析分别发现了杂合 COL4A5 缺失和 HNF1B 重复/缺失。总的来说,在所选择的个体中,致病性变异的存在率为 12.5%(34/271),意义不明的变异存在率为 9.6%(26/271)。生物信息学预测与 MUC1(SNaPshot)的金标准诊断相结合,无法在任何个体中识别出典型的胞嘧啶重复(“c.428dupC”),这意味着 ADTKD-MUC1 很罕见。我们的研究表明,在所选择的个体中,>10%的个体携带部分与肾小管间质性肾脏疾病相关的致病基因变异。COL4A3/4/5 基因构成最大的部分,这意味着它们经常被临床阿尔波特综合征标准所忽略,并显示出表型的存在。我们发现了一些 ES 管道容易错过的变异。应用的临床过滤标准富集了潜在的遗传疾病。