Vanderbilt Kennedy Center, Vanderbilt University Medical Center, Nashville, Tennessee, United States of America.
Department of Pediatrics, Vanderbilt University Medical Center, Nashville, Tennessee, United States of America.
PLoS One. 2022 Oct 12;17(10):e0266861. doi: 10.1371/journal.pone.0266861. eCollection 2022.
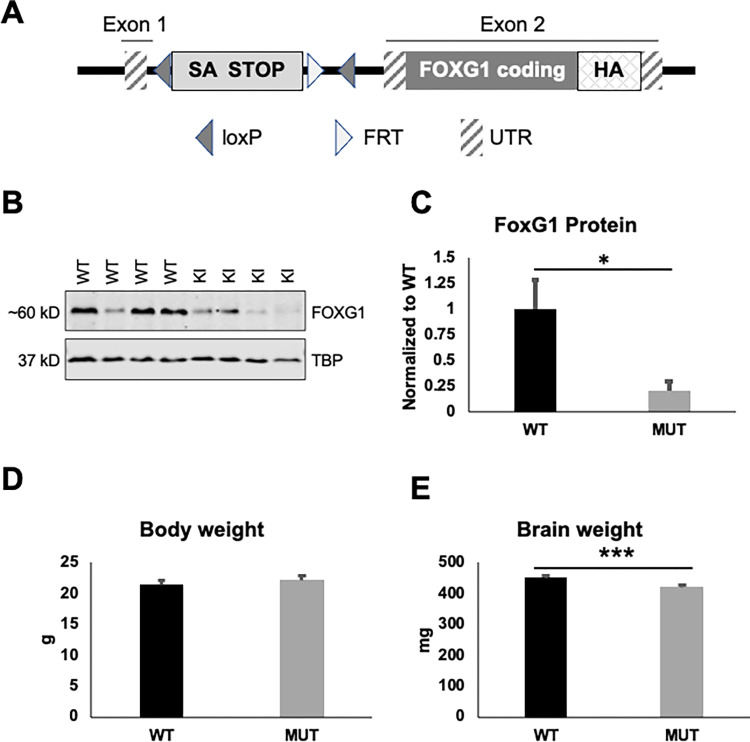
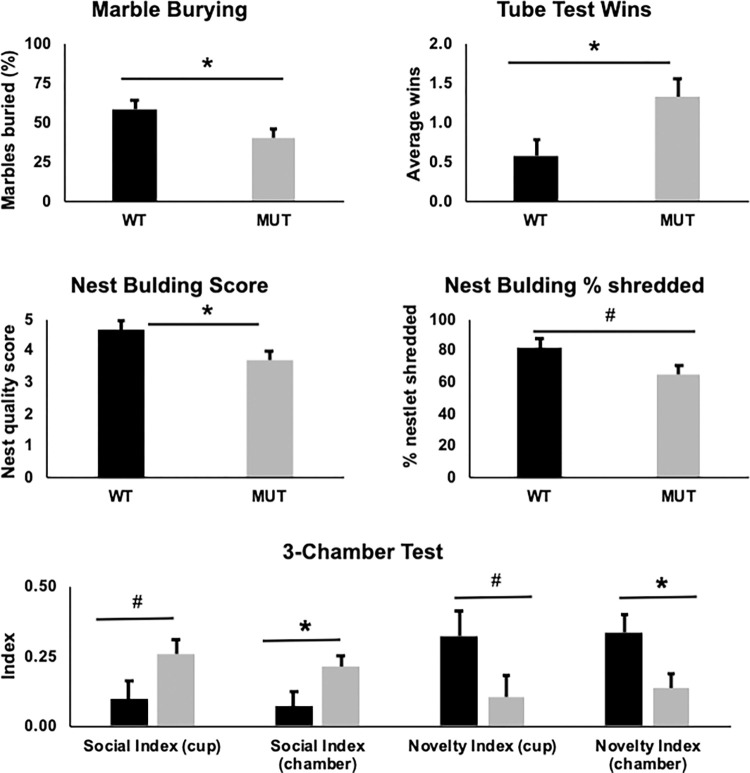
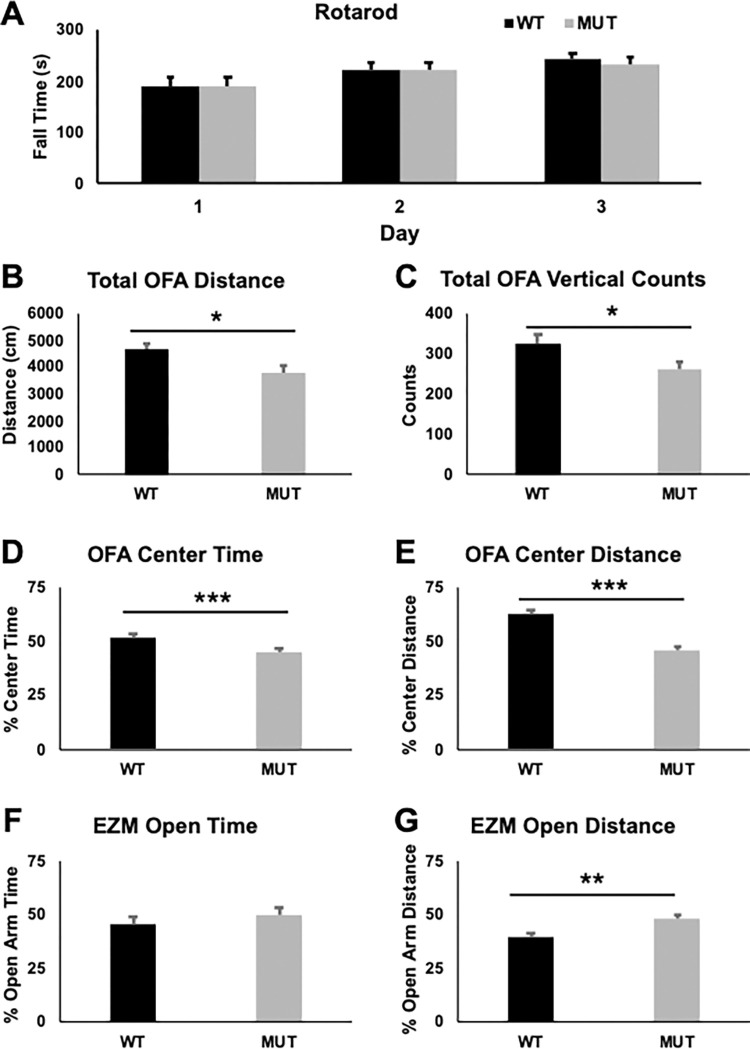
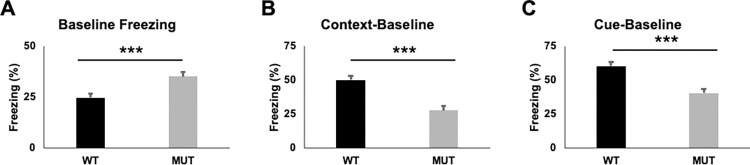
FOXG1 Syndrome (FS) is a devastating neurodevelopmental disorder that is caused by a heterozygous loss-of-function (LOF) mutation of the FOXG1 gene, which encodes a transcriptional regulator important for telencephalic brain development. People with FS have marked developmental delays, impaired ambulation, movement disorders, seizures, and behavior abnormalities including autistic features. Current therapeutic approaches are entirely symptomatic, however the ability to rescue phenotypes in mouse models of other genetic neurodevelopmental disorders such as Rett syndrome, Angelman syndrome, and Phelan-McDermid syndrome by postnatal expression of gene products has led to hope that similar approaches could help modify the disease course in other neurodevelopmental disorders such as FS. While FoxG1 protein function plays a critical role in embryonic brain development, the ongoing adult expression of FoxG1 and behavioral phenotypes that present when FoxG1 function is removed postnatally provides support for opportunity for improvement with postnatal treatment. Here we generated a new mouse allele of Foxg1 that disrupts protein expression and characterized the behavioral and structural brain phenotypes in heterozygous mutant animals. These mutant animals display changes in locomotor behavior, gait, anxiety, social interaction, aggression, and learning and memory compared to littermate controls. Additionally, they have structural brain abnormalities reminiscent of people with FS. This information provides a framework for future studies to evaluate the potential for post-natal expression of FoxG1 to modify the disease course in this severe neurodevelopmental disorder.
FOXG1 综合征(FS)是一种破坏性的神经发育障碍,由 FOXG1 基因的杂合性功能丧失(LOF)突变引起,该基因编码对端脑脑发育很重要的转录调节剂。FS 患者存在明显的发育迟缓、行动障碍、运动障碍、癫痫发作和行为异常,包括自闭症特征。目前的治疗方法完全是对症治疗,然而,通过在其他遗传神经发育障碍(如雷特综合征、天使综合征和 Phelan-McDermid 综合征)的小鼠模型中,通过产后表达基因产物来挽救表型的能力,使得人们希望类似的方法可以帮助改善其他神经发育障碍(如 FS)的疾病进程。虽然 FoxG1 蛋白功能在胚胎大脑发育中起着关键作用,但 FoxG1 的持续成年表达以及 FoxG1 功能被去除后出现的行为表型为产后治疗提供了改善的机会。在这里,我们生成了一种新的 Foxg1 小鼠等位基因,该基因破坏了蛋白质的表达,并对杂合突变动物的行为和大脑结构表型进行了特征描述。与同窝对照相比,这些突变动物的运动行为、步态、焦虑、社交互动、攻击行为以及学习和记忆能力发生了变化。此外,它们还存在类似于 FS 患者的大脑结构异常。这些信息为未来的研究提供了一个框架,以评估产后表达 FoxG1 修饰这种严重神经发育障碍疾病进程的潜力。