Wellcome-MRC Cambridge Stem Cell Institute, University of Cambridge, Jeffrey Cheah Biomedical Centre, Puddicombe Way, Cambridge Biomedical Campus, Cambridge CB2 0AW, UK.
MRC Human Genetics Unit, MRC Institute of Genetics and Cancer, The University of Edinburgh, Western General Hospital, Edinburgh EH4 2XU, UK.
Stem Cell Reports. 2023 Jan 10;18(1):47-63. doi: 10.1016/j.stemcr.2022.09.007. Epub 2022 Oct 13.
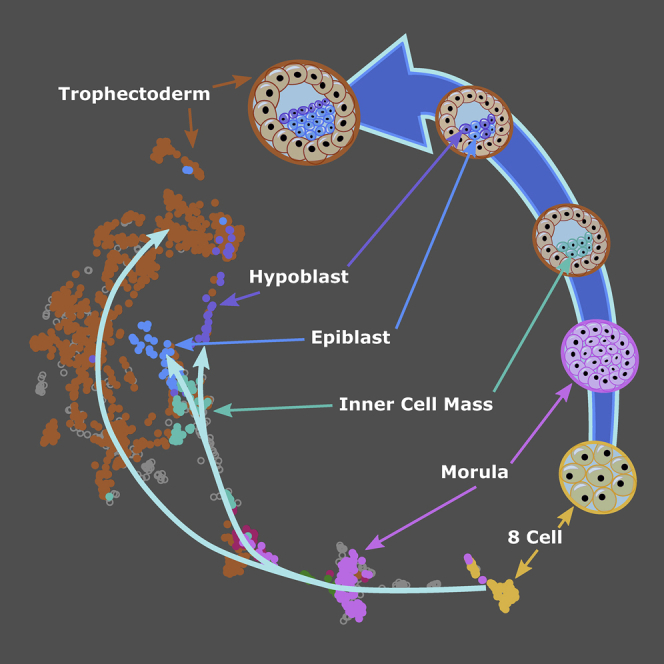
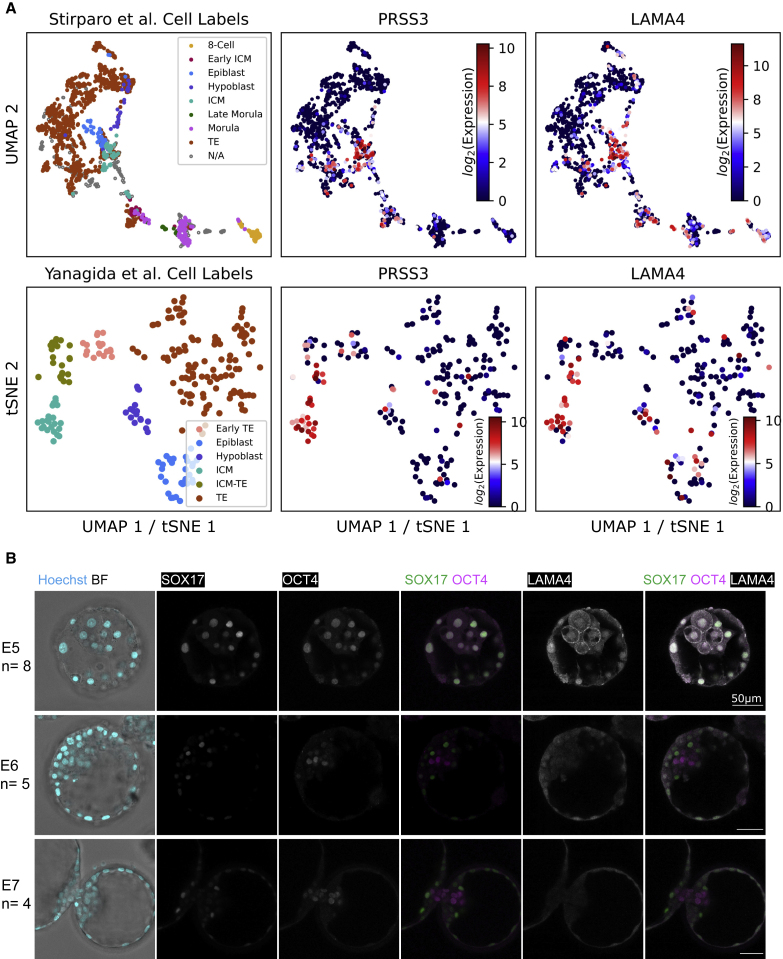
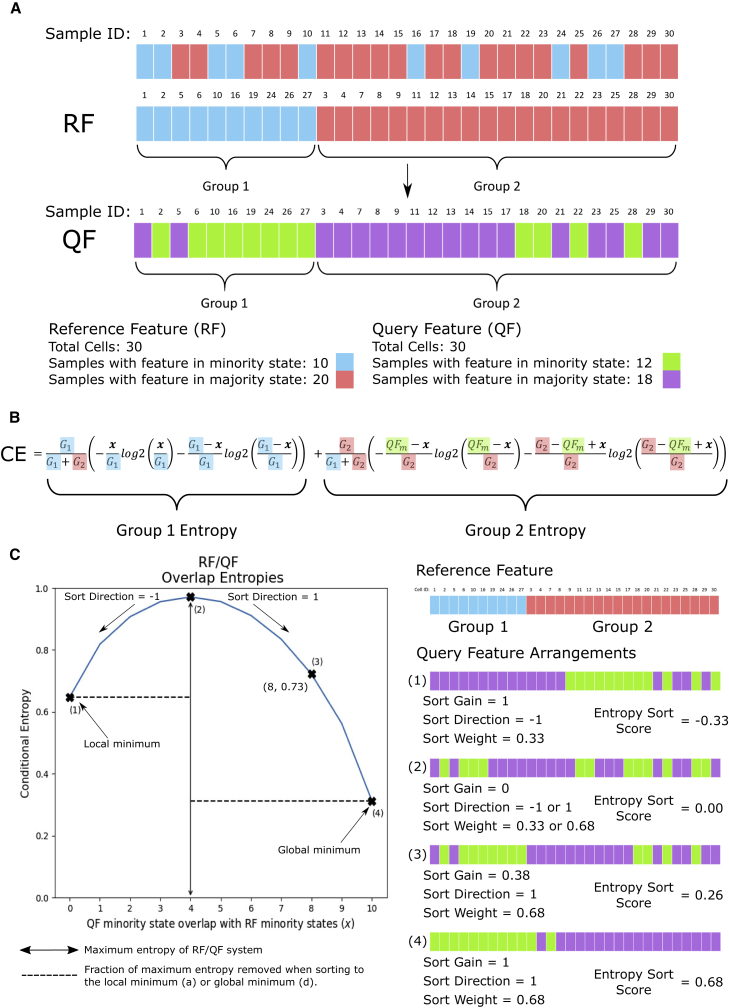
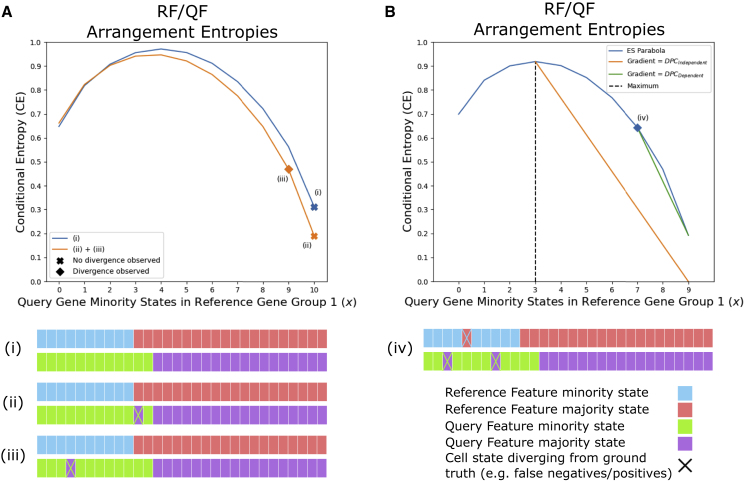
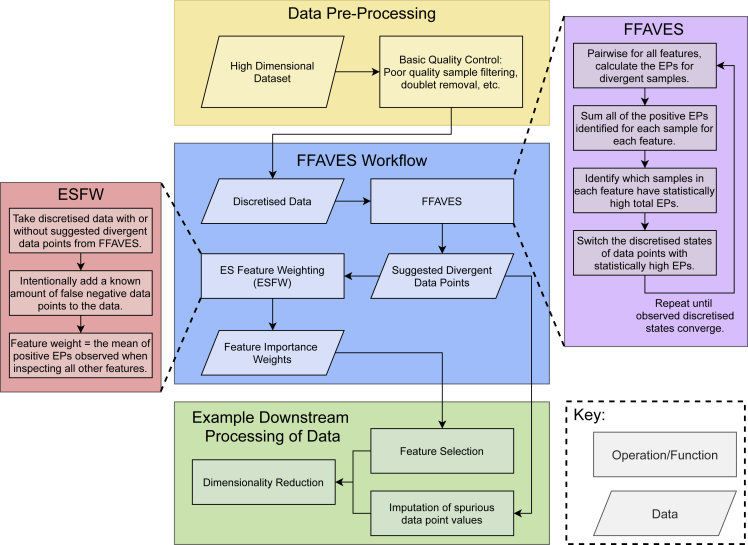
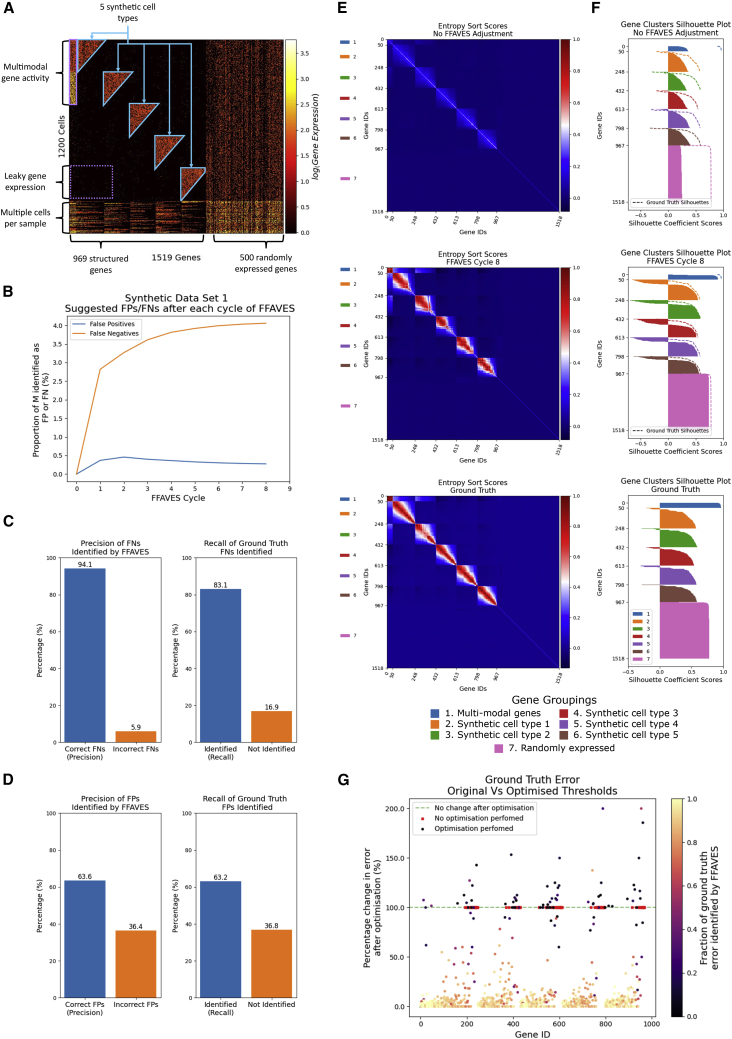
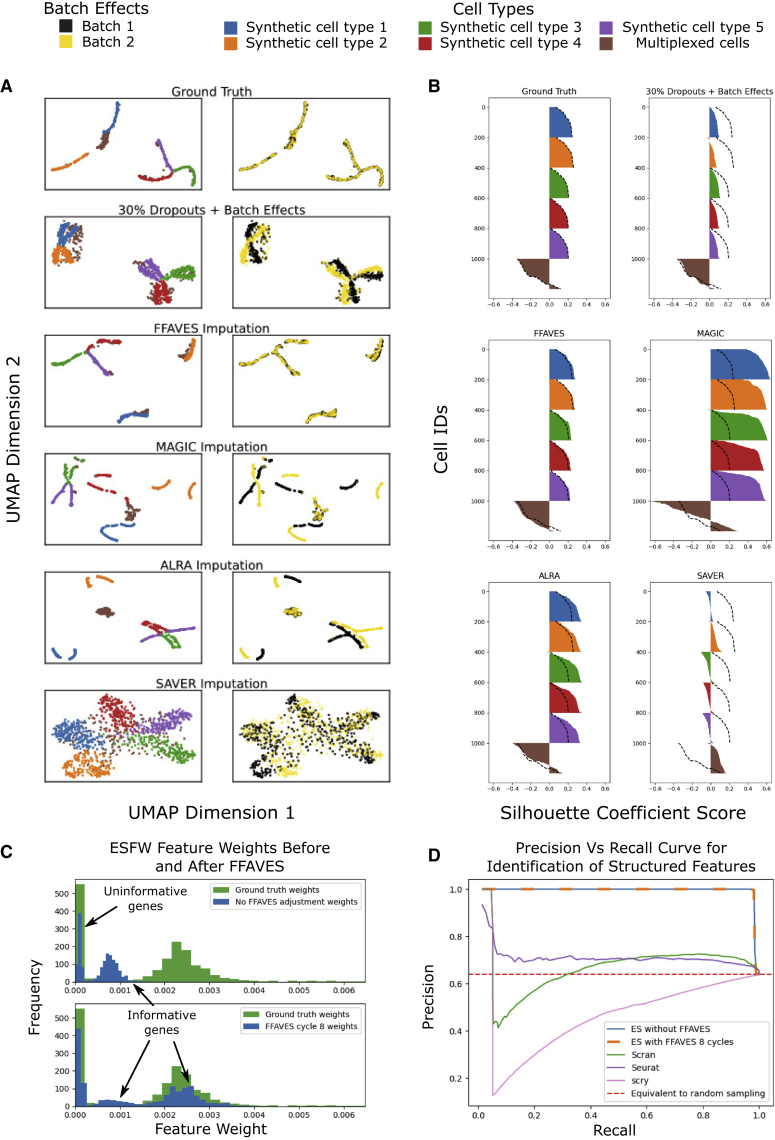
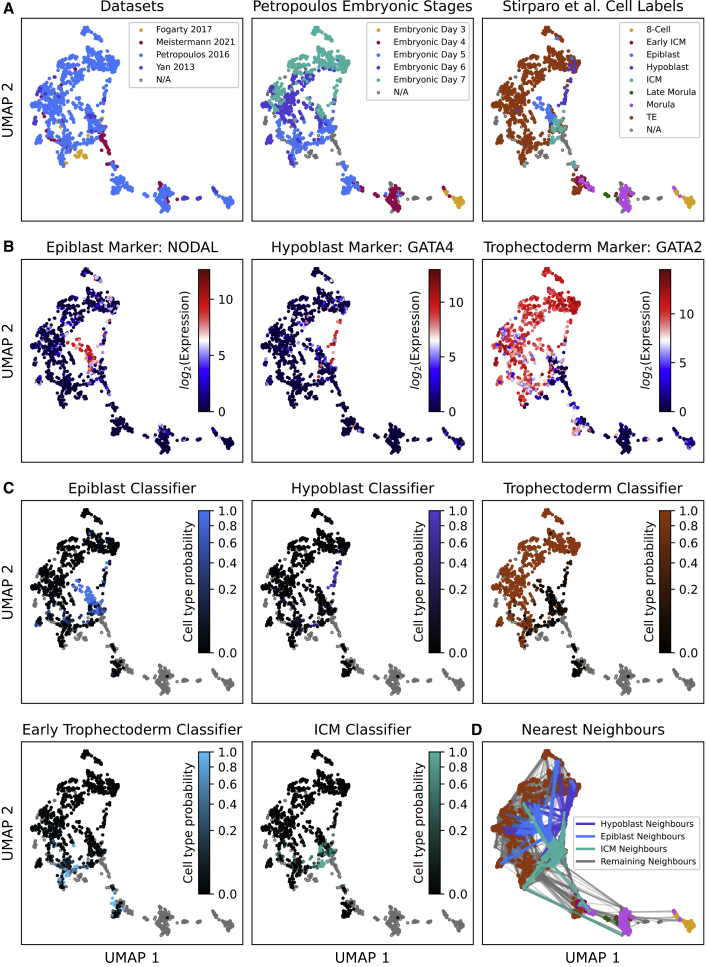
A major challenge in single-cell gene expression analysis is to discern meaningful cellular heterogeneity from technical or biological noise. To address this challenge, we present entropy sorting (ES), a mathematical framework that distinguishes genes indicative of cell identity. ES achieves this in an unsupervised manner by quantifying if observed correlations between features are more likely to have occurred due to random chance versus a dependent relationship, without the need for any user-defined significance threshold. On synthetic data, we demonstrate the removal of noisy signals to reveal a higher resolution of gene expression patterns than commonly used feature selection methods. We then apply ES to human pre-implantation embryo single-cell RNA sequencing (scRNA-seq) data. Previous studies failed to unambiguously identify early inner cell mass (ICM), suggesting that the human embryo may diverge from the mouse paradigm. In contrast, ES resolves the ICM and reveals sequential lineage bifurcations as in the classical model. ES thus provides a powerful approach for maximizing information extraction from high-dimensional datasets such as scRNA-seq data.
单细胞基因表达分析的一个主要挑战是从技术或生物学噪声中辨别有意义的细胞异质性。为了解决这个挑战,我们提出了熵排序(ES),这是一个数学框架,可以区分指示细胞身份的基因。ES 通过量化观察到的特征之间的相关性是更有可能由于随机机会而发生,还是由于依赖关系而发生,而无需任何用户定义的显著性阈值,从而以无监督的方式实现这一点。在合成数据上,我们证明了去除噪声信号可以揭示比常用特征选择方法更高分辨率的基因表达模式。然后,我们将 ES 应用于人类植入前胚胎单细胞 RNA 测序 (scRNA-seq) 数据。以前的研究未能明确识别早期内细胞团 (ICM),这表明人类胚胎可能与小鼠模型不同。相比之下,ES 解决了 ICM 问题,并揭示了与经典模型中类似的顺序谱系分支。因此,ES 为从 scRNA-seq 等高维数据集提取信息提供了一种强大的方法。