Departments of Surgery, University of Missouri School of Medicine, Columbia, MO, USA.
Departments of Surgery, University of Missouri School of Medicine, Columbia, MO, USA; Department of Medical Pharmacology & Physiology, University of Missouri School of Medicine, Columbia, MO, USA.
Redox Biol. 2022 Dec;58:102524. doi: 10.1016/j.redox.2022.102524. Epub 2022 Oct 28.
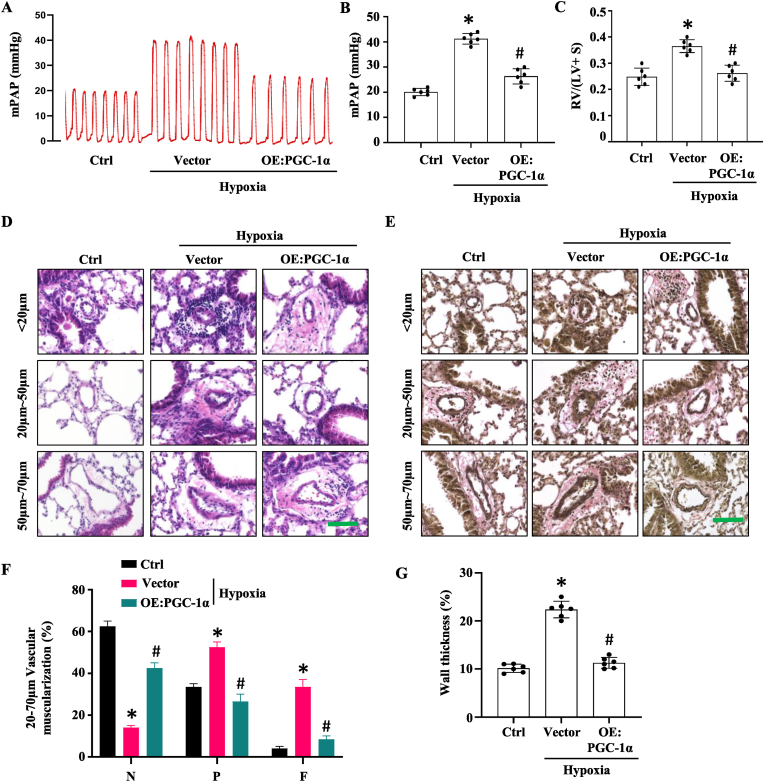
Pulmonary hypertension (PH) induced by chronic hypoxia is characterized by thickening of pulmonary artery walls, elevated pulmonary vascular resistance, and right-heart failure. Dysfunction of endothelial cells is the hallmark event in the progression of PH. Among various mechanisms, endothelial to mesenchymal transition (EndoMT) has emerged as an important source of endothelial cell dysfunction in PH. However, the mechanisms underlying the EndoMT in PH remain largely unknown. Our results showed that peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) expression was decreased in pulmonary arterial endothelial cells (PAECs) in PH patients and hypoxia-induced PH mouse model compared to the normal controls. Endothelial-specific overexpression of PGC-1α using nanoparticle delivery significantly attenuated the progression of PH, as shown by the significantly decreased right ventricular systolic pressure and diminished artery thickness as well as reduced vascular muscularization. Moreover, Endothelial-specific overexpression of PGC-1α blocked the EndoMT of PAECs during PH, indicating that loss of PGC-1α promotes PH development by mediating EndoMT, which damages the integrity of endothelium. Intriguingly, we found that PGC-1α overexpression rescued the expression of endothelial nitric oxide synthase in mouse lung tissues that was deceased by hypoxia treatment in vivo and in endothelial cells treated with TGF-β in vitro. Consistently, PAECs and vascular smooth muscle co-culture showed that overexpression of PGC-1α in PAECs increases nitric oxide release, which would likely diffuse to smooth muscle cells, where it activates specific protein kinases, and initiates SMC relaxation by diminishing the calcium flux. Endothelial-specific overexpression of PGC-1α also attenuated hypoxia-induced pulmonary artery stiffness which appeared to be caused by both the decreased endothelial nitric oxide production and increased vascular remodeling. Taken together, these results demonstrated that endothelial-specific delivery of PGC-1α prevents PH development by inhibiting EndoMT of PAECs and thus restoring endothelial function and reducing vascular remodeling.
慢性缺氧引起的肺动脉高压(PH)的特征是肺血管壁增厚、肺血管阻力升高和右心衰竭。内皮细胞功能障碍是 PH 进展的标志性事件。在各种机制中,内皮向间充质转化(EndoMT)已成为 PH 中内皮细胞功能障碍的重要来源。然而,PH 中 EndoMT 的机制在很大程度上仍然未知。我们的结果表明,与正常对照组相比,PH 患者和缺氧诱导的 PH 小鼠模型中的肺动脉内皮细胞(PAEC)中过氧化物酶体增殖物激活受体γ共激活因子 1-α(PGC-1α)的表达降低。通过纳米颗粒递送在内皮细胞中特异性过表达 PGC-1α 可显著减轻 PH 的进展,表现为右心室收缩压显著降低,动脉厚度减小以及血管肌化减少。此外,内皮细胞特异性过表达 PGC-1α 可阻断 PH 期间的 PAEC 发生 EndoMT,表明 PGC-1α 的缺失通过介导 EndoMT 促进 PH 的发展,从而破坏内皮的完整性。有趣的是,我们发现 PGC-1α 过表达可恢复体内缺氧处理和体外 TGF-β处理的内皮细胞中表达的内皮型一氧化氮合酶。一致地,PAEC 和血管平滑肌共培养显示,PAEC 中 PGC-1α 的过表达增加了一氧化氮的释放,一氧化氮可能扩散到平滑肌细胞,在平滑肌细胞中,它通过减少钙流来激活特定的蛋白激酶,并启动 SMC 松弛。内皮细胞特异性过表达 PGC-1α 还可减轻缺氧诱导的肺动脉僵硬,这似乎是由于内皮一氧化氮产生减少和血管重塑增加所致。总之,这些结果表明,内皮细胞特异性递送 PGC-1α 通过抑制 PAEC 的 EndoMT 来预防 PH 的发展,从而恢复内皮功能并减少血管重塑。