Univ Angers, Nantes Université, CHU Angers, Inserm, CNRS, CRCI2NA, Angers, France.
Department of Laboratory Medicine, Medical University of Vienna, Vienna, Austria.
Blood. 2023 Apr 20;141(16):1909-1921. doi: 10.1182/blood.2022017578.
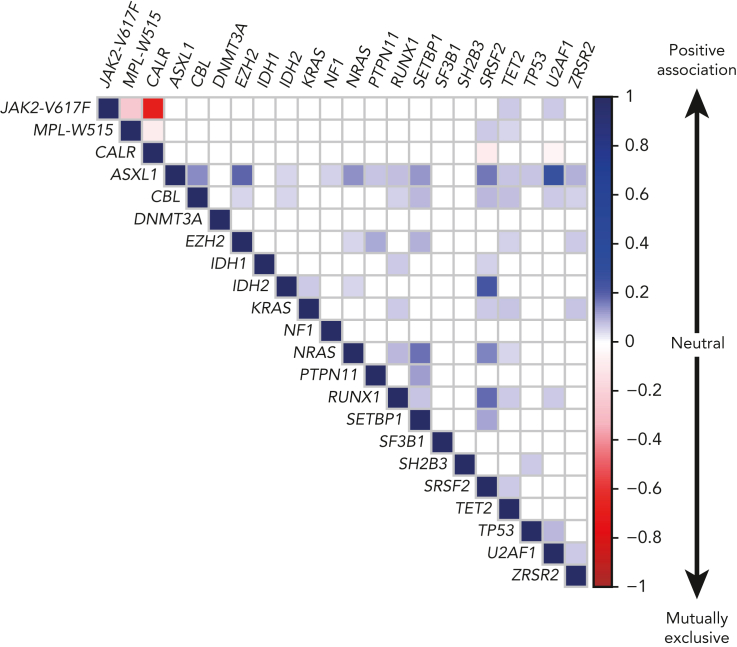
:ABL1-negative myeloproliferative neoplasms (MPNs) are clonal diseases originating from a single hematopoietic stem cell that cause excessive production of mature blood cells. The 3 subtypes, that is, polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF), are diagnosed according to the World Health Organization (WHO) and international consensus classification (ICC) criteria. Acquired gain-of-function mutations in 1 of 3 disease driver genes (JAK2, CALR, and MPL) are the causative events that can alone initiate and promote MPN disease without requiring additional cooperating mutations. JAK2-p.V617F is present in >95% of PV patients, and also in about half of the patients with ET or PMF. ET and PMF are also caused by mutations in CALR or MPL. In ∼10% of MPN patients, those referred to as being "triple negative," none of the known driver gene mutations can be detected. The common theme between the 3 driver gene mutations and triple-negative MPN is that the Janus kinase-signal transducer and activator of transcription (JAK/STAT) signaling pathway is constitutively activated. We review the recent advances in our understanding of the early events after the acquisition of a driver gene mutation. The limiting factor that determines the frequency at which MPN disease develops with a long latency is not the acquisition of driver gene mutations, but rather the expansion of the clone. Factors that control the conversion from clonal hematopoiesis to MPN disease include inherited predisposition, presence of additional mutations, and inflammation. The full extent of knowledge of the mutational landscape in individual MPN patients is now increasingly being used to predict outcome and chose the optimal therapy.
:ABL1 阴性骨髓增殖性肿瘤(MPN)是起源于单个造血干细胞的克隆性疾病,导致成熟血细胞过度生成。根据世界卫生组织(WHO)和国际共识分类(ICC)标准,将 3 种亚型,即真性红细胞增多症(PV)、特发性血小板增多症(ET)和原发性骨髓纤维化(PMF),进行诊断。在 3 种疾病驱动基因(JAK2、CALR 和 MPL)中的 1 种获得功能获得性突变是导致疾病的原因,它可以单独引发和促进 MPN 疾病,而不需要其他协同突变。JAK2-p.V617F 存在于 >95%的 PV 患者中,也存在于约一半的 ET 或 PMF 患者中。ET 和 PMF 也由 CALR 或 MPL 的突变引起。在约 10%的 MPN 患者中,那些被称为“三阴性”的患者,无法检测到已知的驱动基因突变。这 3 种驱动基因突变和三阴性 MPN 的共同主题是,Janus 激酶-信号转导和转录激活因子(JAK/STAT)信号通路被持续激活。我们回顾了对获得驱动基因突变后早期事件的理解的最新进展。决定 MPN 疾病具有长潜伏期的频率的限制因素不是驱动基因突变的获得,而是克隆的扩展。控制从克隆性造血到 MPN 疾病的转化的因素包括遗传易感性、额外突变的存在和炎症。个体 MPN 患者的突变景观的全部知识现在越来越多地用于预测结果和选择最佳治疗方法。