Bukavina Laura, Prunty Megan, Isali Ilaha, Calaway Adam, Ginwala Rashida, Sindhani Mohit, Ghannoum Mahmoud, Mishra Kirtishri, Kutikov Alexander, Uzzo Robert G, Ponsky Lee E, Abbosh Philip H
Department of Urology, University Hospitals, Cleveland, OH, USA.
Case Comprehensive Cancer Center, Cleveland, OH, USA.
Eur Urol Open Sci. 2022 Jul 12;43:5-13. doi: 10.1016/j.euros.2022.06.005. eCollection 2022 Sep.
Until recently, the properties of microbiome and mycobiome in humans and its relevance to disease have largely been unexplored. While the interest of microbiome and malignancy over the past few years have burgeoned with advent of new technologies, no research describing the composition of mycobiome in bladder cancer has been done. Deciphering of the metagenome and its aggregate genetic information can be used to understand the functional properties and relationships between the bacteria, fungi, and cancer.
The aim of this project is to characterize the compositional range of the normal versus bladder cancer mycobiome of the gut.
An internal transcribed spacer (ITS) survey of 52 fecal samples was performed to evaluate the gut mycobiome differences between noncancer controls and bladder cancer patients.
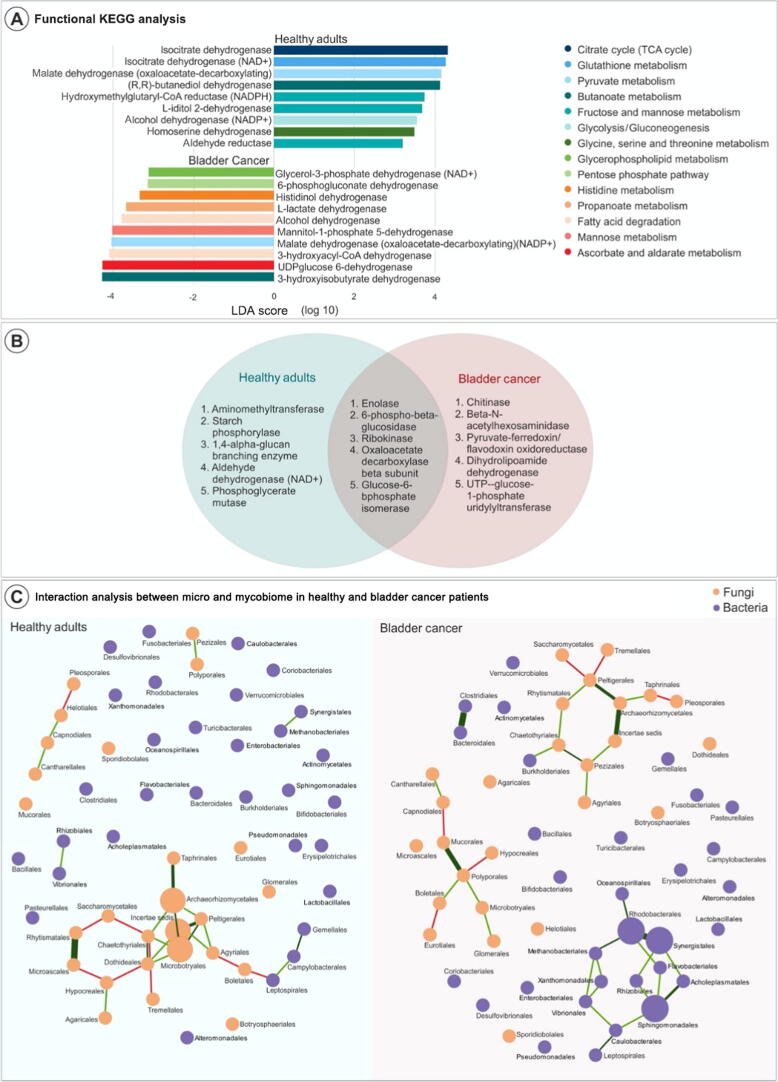
Our study evaluated the differences in mycobiome among patients with bladder cancer, versus matched controls. Our secondary analysis evaluated compositional differences in the gut as a function of response status with neoadjuvant chemotherapy. Data demultiplexing and classification were performed using the QIIME v.1.1.1.1 platform. The Ion Torrent-generated fungal ITS sequence data were processed using QIIME (v.1.9.1), and the reads were demultiplexed, quality filtered, and clustered into operation taxonomic units using default parameters. Alpha and beta diversity were computed and plotted in Phyloseq, principal coordinate analysis was performed on Bray-Curtis dissimilarity indices, and a one-way permutational multivariate analysis of variance was used to test for significant differences between cohorts. Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) was applied to infer functional categories associated with taxonomic composition.
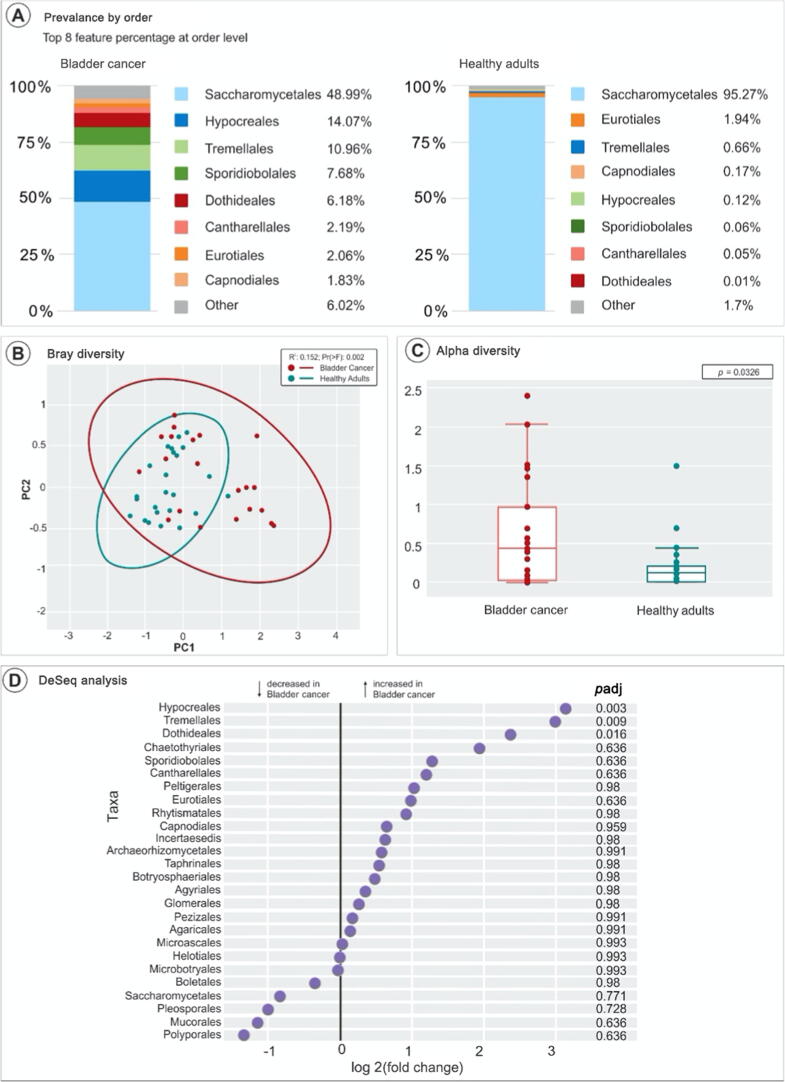
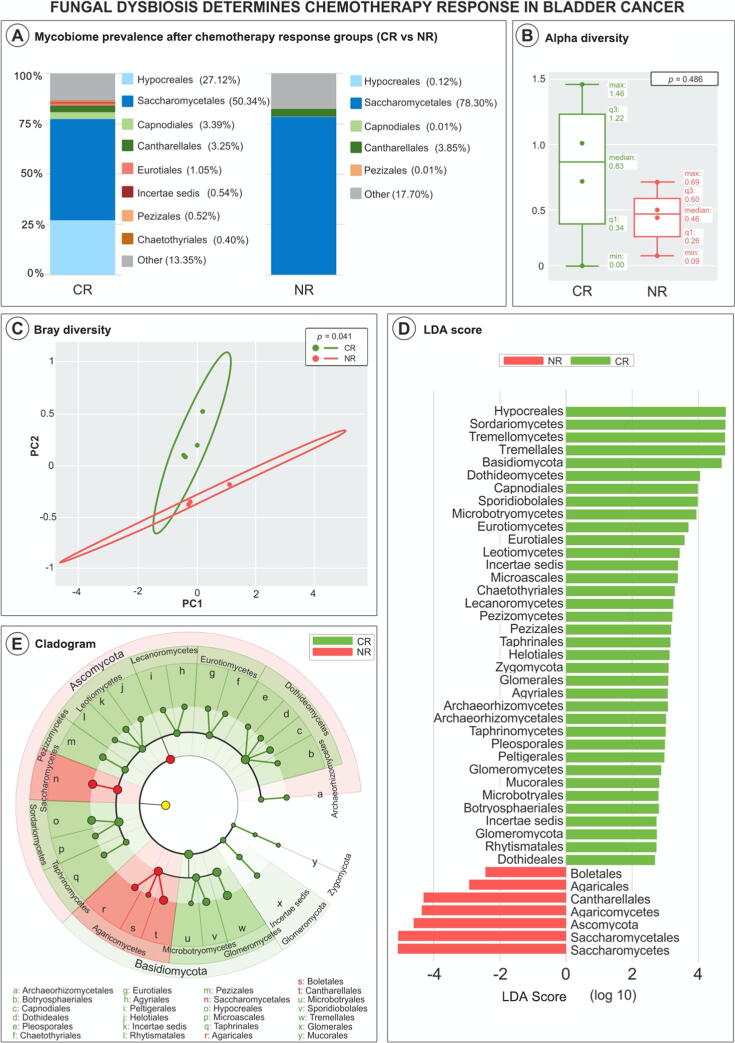
We found distinctive mycobiome differences between control group ( = 32) and bladder cancer ( = 29) gut flora, and identified an increasing abundance of , , and . Significant differences in alpha and beta diversity were present between the groups (control vs bladder; = 0.002), noting distinct compositions within each cohort. A subgroup analysis by sex and neoadjuvant chemotherapy status did not show any further differences in mycobiome composition and diversity. Our results indicate that the gut mycobiome may modulate tumor response to preoperative chemotherapy in bladder cancer patients. We propose that patients with a "favorable" mycobiome composition (eg, high diversity, and low abundance of and ) may have enhanced systemic immune response to chemotherapy through antigen presentation.
Our study is the first to characterize the enteric mycobiome in patients with bladder cancer and describe complex ecological network alterations, indicating complex bacteria-fungi interactions, particularly highlighted among patients with complete neoadjuvant chemotherapy response.
Our study has demonstrated that the composition of stool mycobiome (fungal inhabitants of the gastrointestinal tract) in patients with bladder cancer is different from that in noncancer individuals. Furthermore, when evaluating how patients respond to chemotherapy given prior to their surgery, our study noted significant differences between patients who responded and those who did not.
直到最近,人类微生物组和真菌组的特性及其与疾病的相关性在很大程度上仍未得到探索。尽管随着新技术的出现,过去几年微生物组与恶性肿瘤之间的研究兴趣迅速增长,但尚未有关于膀胱癌真菌组组成的研究。宏基因组及其聚合遗传信息的解读可用于了解细菌、真菌与癌症之间的功能特性及关系。
本项目旨在表征肠道正常与膀胱癌真菌组的组成范围。
设计、设置与参与者:对52份粪便样本进行内部转录间隔区(ITS)检测,以评估非癌症对照者与膀胱癌患者之间肠道真菌组的差异。
我们的研究评估了膀胱癌患者与匹配对照者之间真菌组的差异。我们的二次分析评估了肠道组成差异与新辅助化疗反应状态的关系。使用QIIME v.1.1.1.1平台进行数据解复用和分类。使用QIIME(v.1.9.1)处理离子 torrent 生成的真菌ITS序列数据,并对读数进行解复用、质量过滤,并使用默认参数聚类为操作分类单元。计算α和β多样性并在Phyloseq中绘制,对Bray-Curtis差异指数进行主坐标分析,并使用单因素置换多元方差分析来检验队列之间的显著差异。应用未观察状态重建的群落系统发育调查(PICRUSt)来推断与分类组成相关的功能类别。
我们发现对照组(n = 32)和膀胱癌(n = 29)肠道菌群之间存在明显的真菌组差异,并确定了[具体菌种名称1]、[具体菌种名称2]和[具体菌种名称3]的丰度增加。两组之间(对照组与膀胱癌组;P = 0.002)的α和β多样性存在显著差异,每个队列内的组成各不相同。按性别和新辅助化疗状态进行的亚组分析未显示真菌组组成和多样性有任何进一步差异。我们的结果表明,肠道真菌组可能调节膀胱癌患者对术前化疗的肿瘤反应。我们提出,具有“有利”真菌组组成(例如,高多样性以及[具体菌种名称1]和[具体菌种名称2]的低丰度)的患者可能通过抗原呈递增强对化疗的全身免疫反应。
我们的研究首次表征了膀胱癌患者的肠道真菌组,并描述了复杂的生态网络改变,表明细菌 - 真菌之间存在复杂的相互作用,在新辅助化疗完全缓解的患者中尤为突出。
我们的研究表明,膀胱癌患者粪便真菌组(胃肠道中的真菌居民)的组成与非癌症个体不同。此外,在评估患者对手术前给予的化疗的反应时,我们的研究注意到有反应和无反应患者之间存在显著差异。