Ifremer, ASIM, F-17390 La Tremblade, France.
IHPE, Univ. Montpellier, CNRS, Ifremer, UPVD, F-34095 Montpellier, France.
Microb Genom. 2022 Nov;8(11). doi: 10.1099/mgen.0.000895.
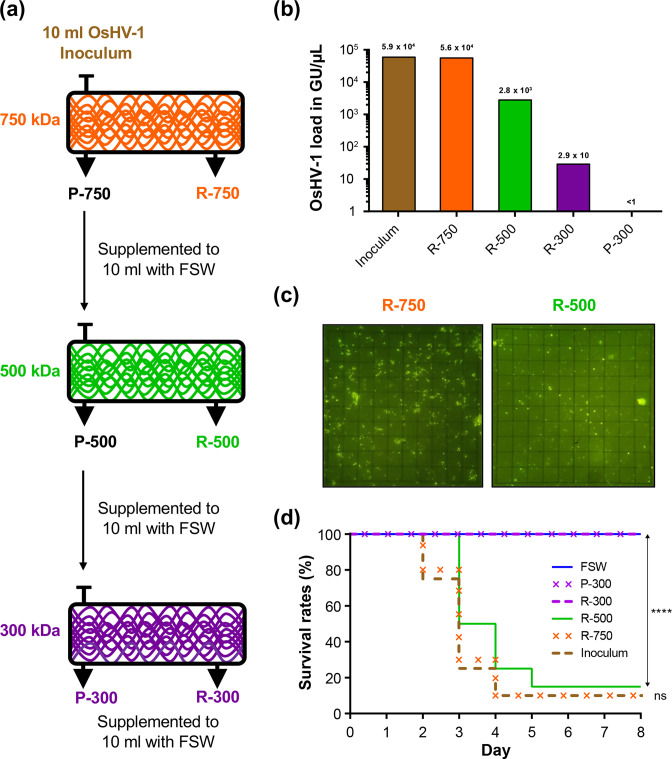
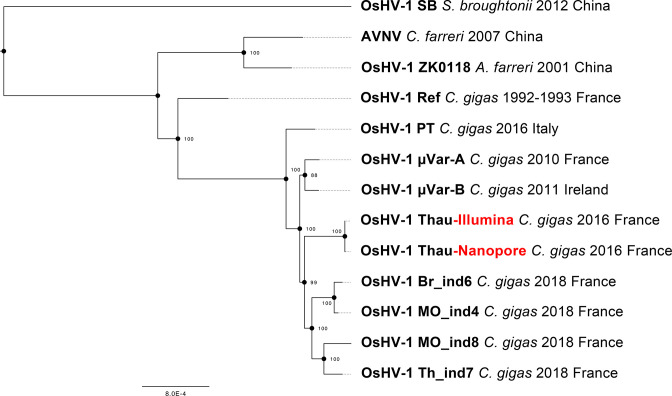
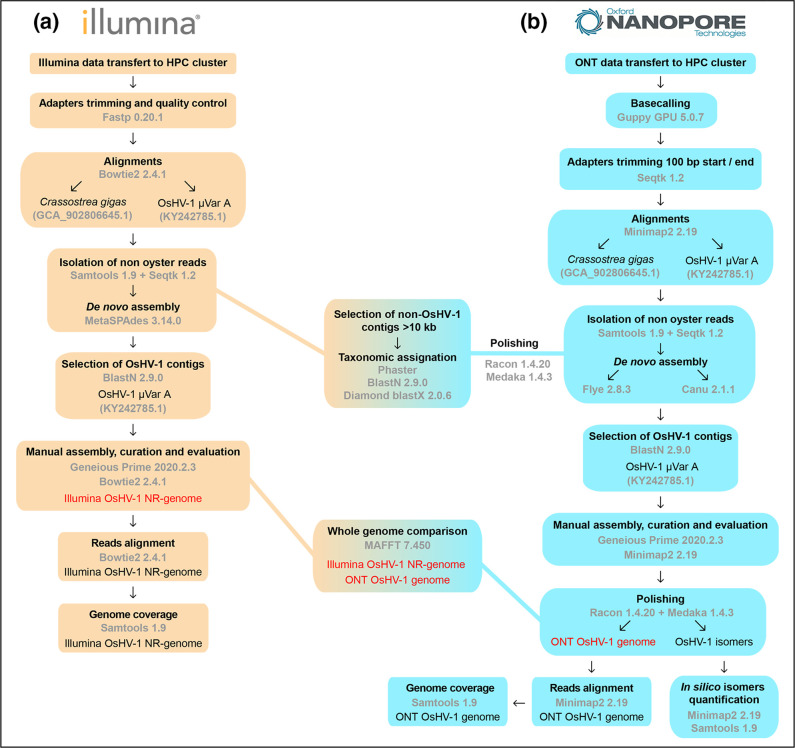
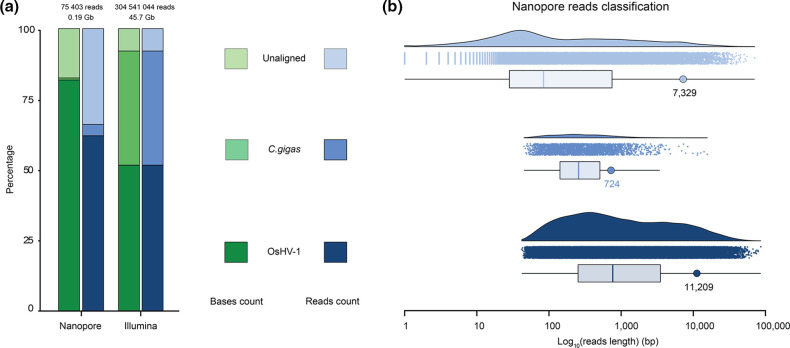
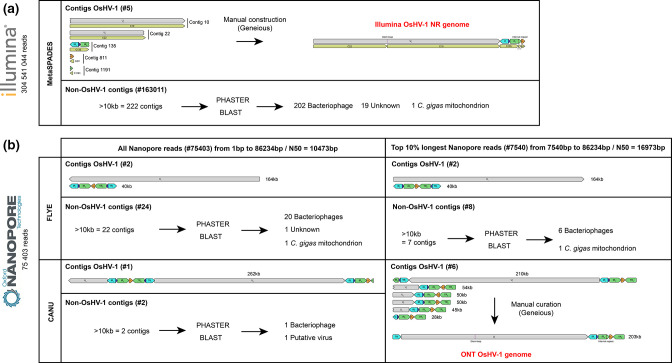
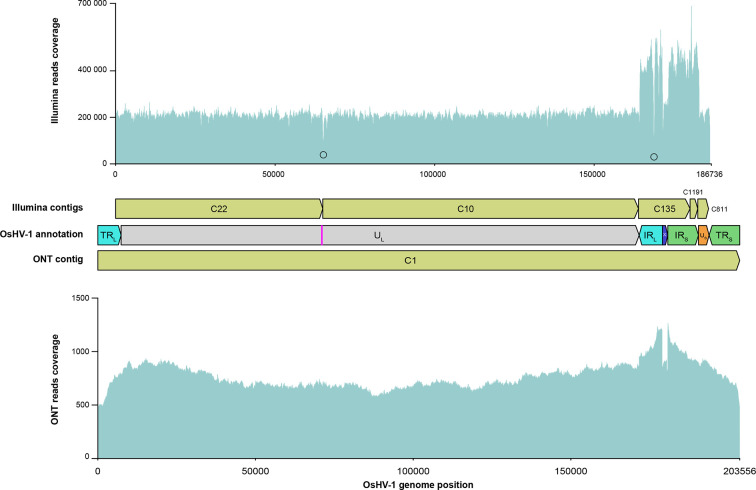
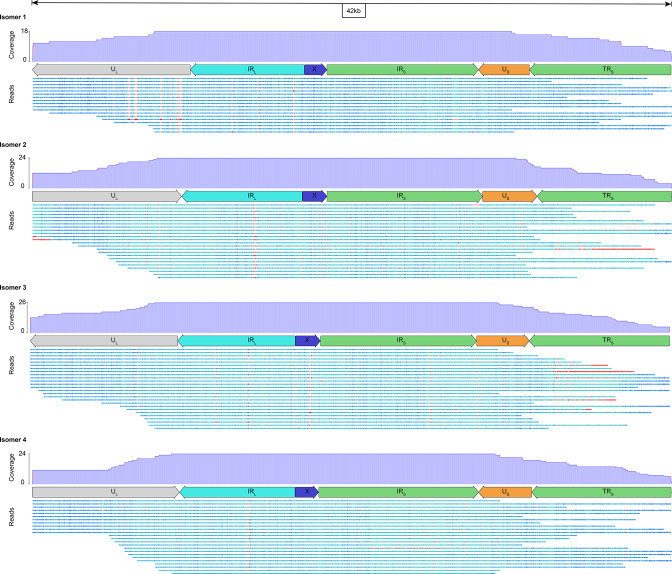
Whole-genome sequencing is widely used to better understand the transmission dynamics, the evolution and the emergence of new variants of viral pathogens. This can bring crucial information to stakeholders for disease management. Unfortunately, aquatic virus genomes are usually difficult to characterize because most of these viruses cannot be easily propagated . Developing methodologies for routine genome sequencing of aquatic viruses is timely given the ongoing threat of disease emergence. This is particularly true for pathogenic viruses infecting species of commercial interest that are widely exchanged between production basins or countries. For example, the ostreid herpesvirus type 1 (OsHV-1) is a Herpesvirus widely associated with mass mortality events of juvenile Pacific oyster . Genomes of Herpesviruses are large and complex with long direct and inverted terminal repeats. In addition, OsHV-1 is unculturable. It therefore accumulates several features that make its genome sequencing and assembly challenging. To overcome these difficulties, we developed a tangential flow filtration (TFF) method to enrich OsHV-1 infective particles from infected host tissues. This virus purification allowed us to extract high molecular weight and high-quality viral DNA that was subjected to Illumina short-read and Nanopore long-read sequencing. Dedicated bioinformatic pipelines were developed to assemble complete OsHV-1 genomes with reads from both sequencing technologies. Nanopore sequencing allowed characterization of new structural variations and major viral isomers while having 99,98 % of nucleotide identity with the Illumina assembled genome. Our study shows that TFF-based purification method, coupled with Nanopore sequencing, is a promising approach to enable in field sequencing of unculturable aquatic DNA virus.
全基因组测序被广泛用于更好地了解病毒病原体的传播动态、进化和新变体的出现。这可以为利益相关者提供疾病管理的关键信息。不幸的是,水生病毒基因组通常难以表征,因为大多数这些病毒不容易繁殖。鉴于疾病不断出现的威胁,开发水生病毒常规基因组测序的方法是及时的。对于感染商业利益物种的致病性病毒来说尤其如此,这些物种在生产盆地或国家之间广泛交换。例如,牡蛎疱疹病毒 1(OsHV-1)是一种广泛与太平洋牡蛎幼体大量死亡事件相关的疱疹病毒。疱疹病毒的基因组较大且复杂,具有长的直接和反向末端重复序列。此外,OsHV-1 是不可培养的。因此,它积累了一些使其基因组测序和组装具有挑战性的特征。为了克服这些困难,我们开发了切向流过滤(TFF)方法,从感染宿主组织中浓缩 OsHV-1 感染颗粒。这种病毒纯化使我们能够提取高分子量和高质量的病毒 DNA,然后对其进行 Illumina 短读和 Nanopore 长读测序。开发了专门的生物信息学管道,以便使用两种测序技术的读取来组装完整的 OsHV-1 基因组。Nanopore 测序允许表征新的结构变异和主要病毒异构体,同时与 Illumina 组装基因组具有 99.98%的核苷酸同一性。我们的研究表明,基于 TFF 的纯化方法与 Nanopore 测序相结合,是一种有前途的方法,可以实现不可培养的水生 DNA 病毒的现场测序。