Zhao Qian, Wang Teng, Wang Huanhuan, Cui Cheng, Zhong Wen, Fu Diyi, Xi Wanlin, Si Lu, Guo Jun, Cheng Ying, Tian Hongqi, Hu Pei
Clinical Pharmacology Research Center, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, State Key Laboratory of Complex Severe and Rare Diseases, NMPA Key Laboratory for Clinical Research and Evaluation of Drug, Beijing Key Laboratory of Clinical PK and PD Investigation for Innovative Drugs, Beijing, China.
Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Department of Renal Cancer and Melanoma, Peking University Cancer Hospital and Research Institute, Beijing, China.
Front Pharmacol. 2022 Nov 1;13:1039416. doi: 10.3389/fphar.2022.1039416. eCollection 2022.
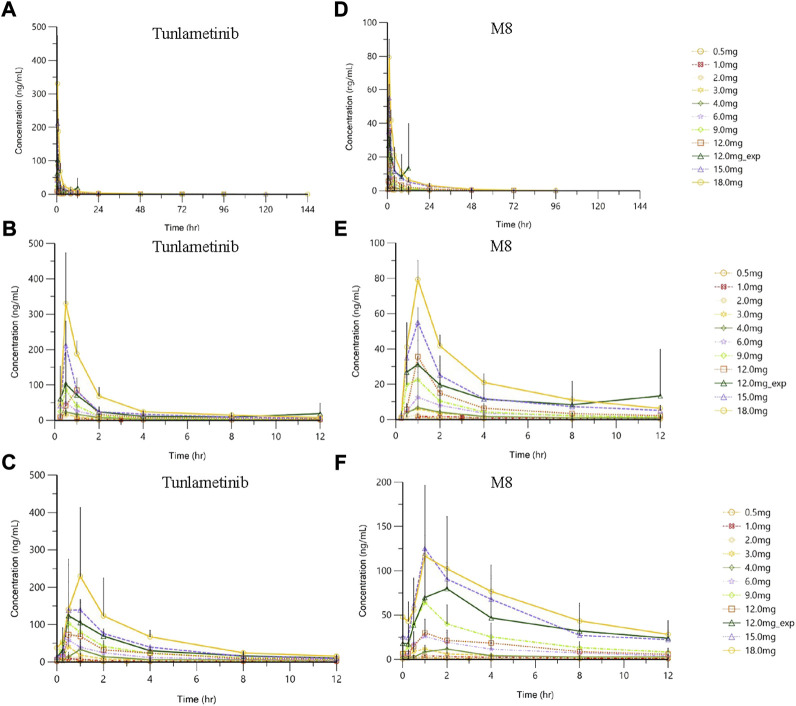
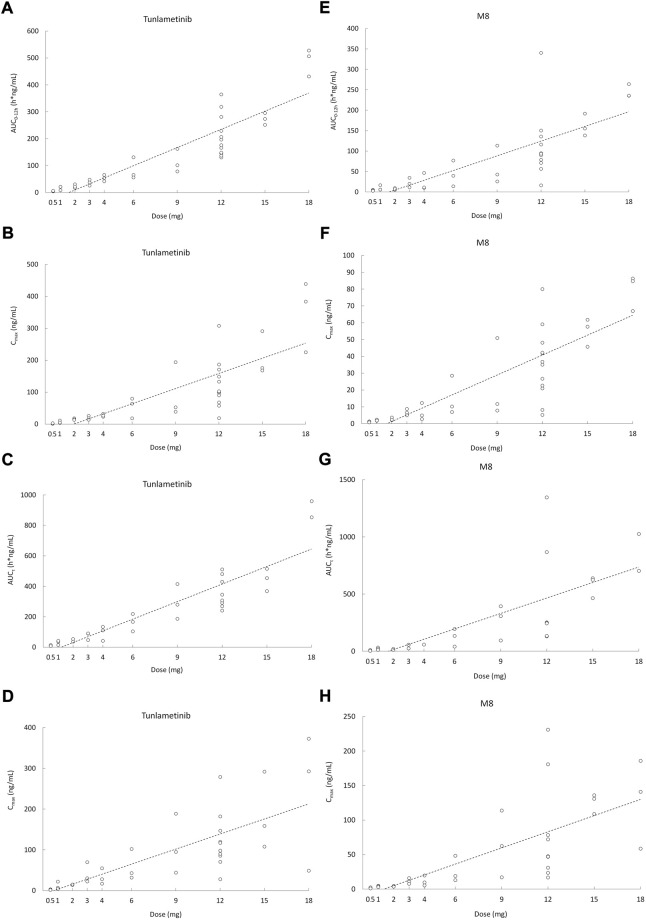
Malignant melanoma is an aggressive disease. Tunlametinib (HL-085) is a potent, selective, and orally bioavailable MEK1/2 inhibitor. The objective of this study was to determine the pharmacokinetics (PK) of tunlametinib and its main metabolite M8 in patients with -mutant melanoma following a single dose and multiple doses in a phase I safety and PK study. A multiple-center phase I study was performed in patients with melanoma including dose-escalation phase and dose-expansion phase. PK following a single oral dose and multiple doses of 0.5-18 mg twice daily was assessed. A total of 30 participants were included in the dose escalation phase and then 11 patients were included in the dose-expansion phase (12 mg twice daily). Tunlametinib plasma concentration rapidly increased after dosing, with a T of 0.5-1 h. Mean elimination half-life (t) was dose-independent and had a range from 21.84 to 34.41 h. Mean apparent clearance (CL/F) and distribution volume (V/F) were 28.44-51.93 L/h and 1199.36-2009.26 L, respectively. The average accumulation ratios of AUC and C after the multiple administration of tunlametinib were 1.64-2.73 and 0.82-2.49, respectively. Tunlametinib was rapidly transformed into the main metabolite M8 and M8 reached the peak concentration about 1 h after administration. Mean t of M8 was 6.1-33.54 h. The body exposure of M8 in plasma was 36%-67% of that of tunlametinib. There were general dose-proportional increases in maximum concentration (C) and area under the curve (AUC) of tunlametinib and M8 both in the single dose phase and in the multiple doses phase. Tunlametinib was absorbed rapidly and eliminated at a medium speed after drug withdrawal. Pharmacokinetic body exposure increased in general dose-proportional manner from 0.5 mg up to 18 mg. Slight accumulation was found after multiple oral doses. The pharmacokinetics of tunlametinib and its metabolite suggest that twice daily dosing is appropriate for tunlametinib.
恶性黑色素瘤是一种侵袭性疾病。图拉替尼(HL-085)是一种强效、选择性且口服生物利用度高的MEK1/2抑制剂。本研究的目的是在一项I期安全性和药代动力学研究中,确定图拉替尼及其主要代谢产物M8在携带 - 突变黑色素瘤患者单剂量和多剂量给药后的药代动力学(PK)。对黑色素瘤患者进行了一项多中心I期研究,包括剂量递增阶段和剂量扩展阶段。评估了单口服剂量和每日两次0.5 - 18 mg多剂量给药后的PK。剂量递增阶段共纳入30名参与者,然后剂量扩展阶段纳入11名患者(每日两次12 mg)。给药后图拉替尼血浆浓度迅速升高,达峰时间(T)为0.5 - 1小时。平均消除半衰期(t)与剂量无关,范围为21.84至34.41小时。平均表观清除率(CL/F)和分布容积(V/F)分别为28.44 - 51.93 L/h和1199.36 - 2009.26 L。图拉替尼多次给药后AUC和C的平均蓄积比分别为1.64 - 2.73和0.82 - 2.49。图拉替尼迅速转化为主要代谢产物M8,M8在给药后约1小时达到峰值浓度。M8的平均t为6.1 - 33.54小时。M8在血浆中的体内暴露量为图拉替尼的36% - 67%。在单剂量阶段和多剂量阶段,图拉替尼和M8的最大浓度(C)和曲线下面积(AUC)均呈现一般剂量比例增加。图拉替尼吸收迅速,停药后以中等速度消除。药代动力学体内暴露量从0.5 mg至18 mg一般呈剂量比例增加。多次口服给药后发现有轻微蓄积。图拉替尼及其代谢产物的药代动力学表明,图拉替尼每日两次给药是合适的。