Centre for Infectious Diseases Control, National Institute for Public Health and the Environment (RIVM), Bilthoven, The Netherlands.
Wageningen Bioveterinary Research (WBVR), Lelystad, The Netherlands.
Transbound Emerg Dis. 2022 Nov;69(6):3881-3895. doi: 10.1111/tbed.14759. Epub 2022 Nov 30.
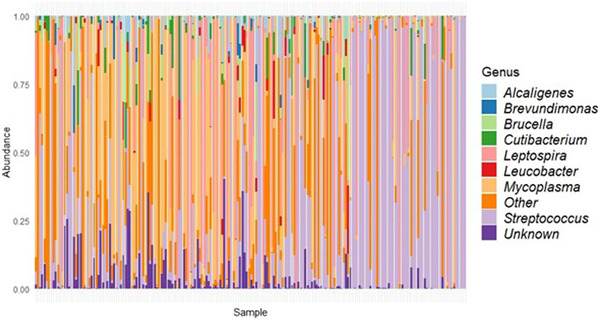
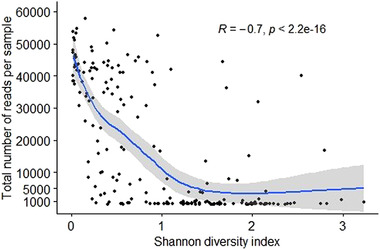
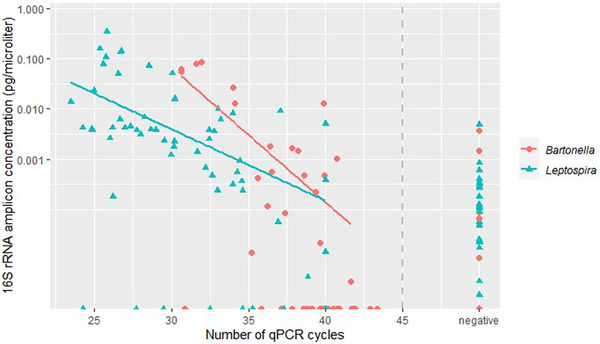
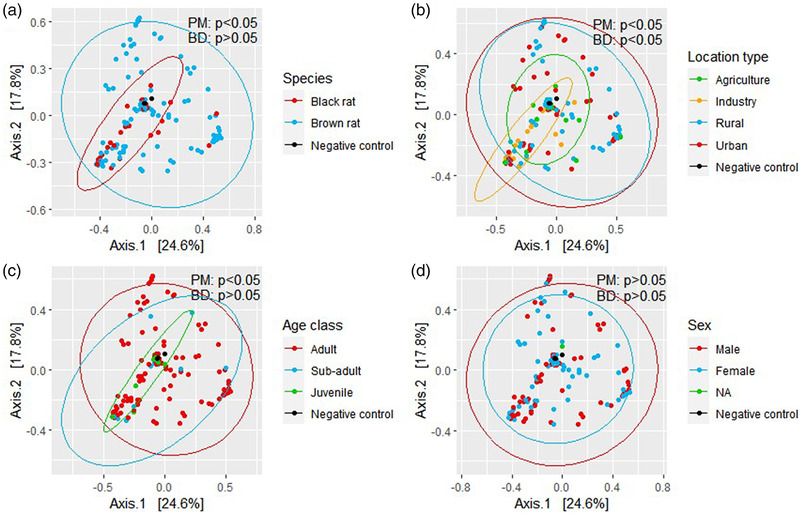
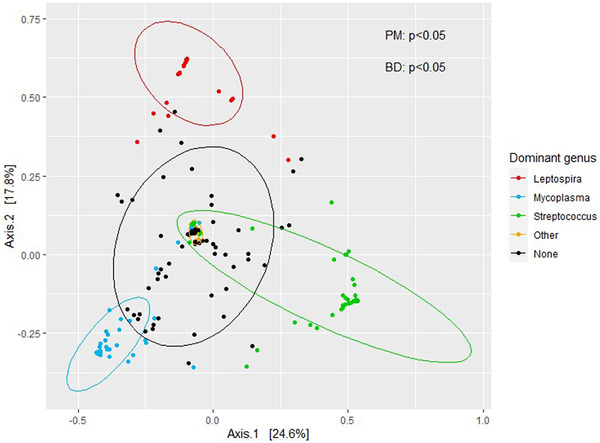
Wild rats can host various zoonotic pathogens. Detection of these pathogens is commonly performed using molecular techniques targeting one or a few specific pathogens. However, this specific way of surveillance could lead to (emerging) zoonotic pathogens staying unnoticed. This problem may be overcome by using broader microbiome-profiling techniques, which enable broad screening of a sample's bacterial or viral composition. In this study, we investigated if 16S rRNA gene amplicon sequencing would be a suitable tool for the detection of zoonotic bacteria in wild rats. Moreover, we used virome-enriched (VirCapSeq) sequencing to detect zoonotic viruses. DNA from kidney samples of 147 wild brown rats (Rattus norvegicus) and 42 black rats (Rattus rattus) was used for 16S rRNA gene amplicon sequencing of the V3-V4 hypervariable region. Blocking primers were developed to reduce the amplification of rat host DNA. The kidney bacterial composition was studied using alpha- and beta-diversity metrics and statistically assessed using PERMANOVA and SIMPER analyses. From the sequencing data, 14 potentially zoonotic bacterial genera were identified from which the presence of zoonotic Leptospira spp. and Bartonella tribocorum was confirmed by (q)PCR or Sanger sequencing. In addition, more than 65% of all samples were dominated (>50% reads) by one of three bacterial taxa: Streptococcus (n = 59), Mycoplasma (n = 39) and Leptospira (n = 25). These taxa also showed the highest contribution to the observed differences in beta diversity. VirCapSeq sequencing in rat liver samples detected the potentially zoonotic rat hepatitis E virus in three rats. Although 16S rRNA gene amplicon sequencing was limited in its capacity for species level identifications and can be more difficult to interpret due to the influence of contaminating sequences in these low microbial biomass samples, we believe it has potential to be a suitable pre-screening method in the future to get a better overview of potentially zoonotic bacteria that are circulating in wildlife.
野生鼠类可以携带各种人畜共患病原体。这些病原体的检测通常使用针对一种或几种特定病原体的分子技术进行。然而,这种特定的监测方式可能会导致(新出现的)人畜共患病原体被忽视。这个问题可以通过使用更广泛的微生物组分析技术来克服,这些技术可以广泛筛选样本的细菌或病毒组成。在这项研究中,我们研究了 16S rRNA 基因扩增子测序是否适合检测野生鼠类中的人畜共患细菌。此外,我们还使用病毒富集(VirCapSeq)测序来检测人畜共患病毒。从 147 只野生褐家鼠(Rattus norvegicus)和 42 只黑家鼠(Rattus rattus)的肾脏样本中提取 DNA,用于扩增子测序 V3-V4 高变区。开发了阻断引物以减少大鼠宿主 DNA 的扩增。使用 alpha 和 beta 多样性指标研究肾脏细菌组成,并使用 PERMANOVA 和 SIMPER 分析进行统计学评估。从测序数据中,鉴定出 14 种可能的人畜共患细菌属,其中通过(q)PCR 或 Sanger 测序证实了人畜共患钩端螺旋体属和博氏疏螺旋体的存在。此外,超过 65%的样本被三种细菌群(链球菌属(n = 59)、支原体属(n = 39)和钩端螺旋体属(n = 25))中的一种占据(>50%的reads)。这些分类群也对观察到的 beta 多样性差异贡献最大。在大鼠肝样 VirCapSeq 测序中,在三只大鼠中检测到了潜在的人畜共患大鼠戊型肝炎病毒。尽管 16S rRNA 基因扩增子测序在种属水平鉴定方面的能力有限,并且由于低微生物生物量样本中污染序列的影响,可能更难解释,但我们相信它具有成为未来适合的预筛选方法的潜力,以更好地了解野生动物中循环的潜在人畜共患细菌。