State Key Laboratory of Oncogenes and Related Genes, Stem Cell Research Center, Shanghai Cancer Institute, Ren Ji Hospital, Shanghai Jiao Tong University School of Medicine, 160 Pujian Rd, Shanghai, 200127, P. R. China.
Department of Biliary-Pancreatic Surgery, Ren Ji Hospital, Shanghai Jiao Tong University School of Medicine, 160 Pujian Rd, Shanghai, 200127, P. R. China.
Adv Sci (Weinh). 2023 Jan;10(2):e2202937. doi: 10.1002/advs.202202937. Epub 2022 Dec 1.
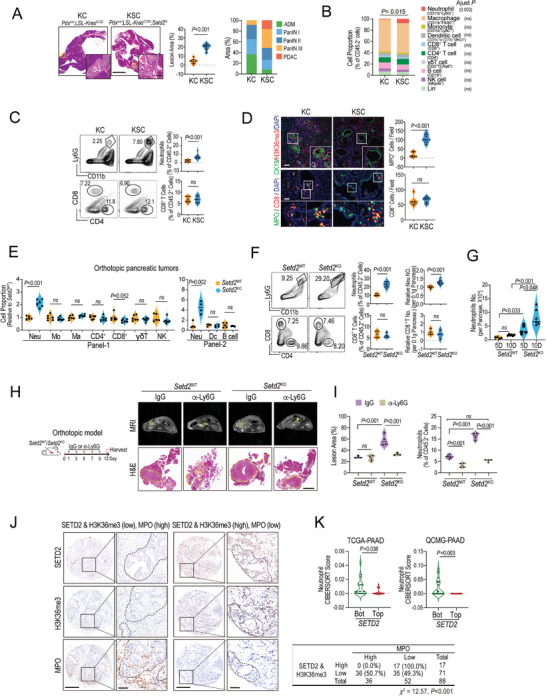
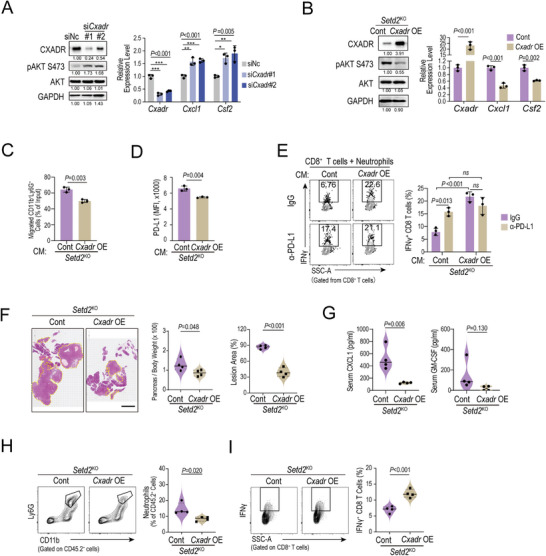
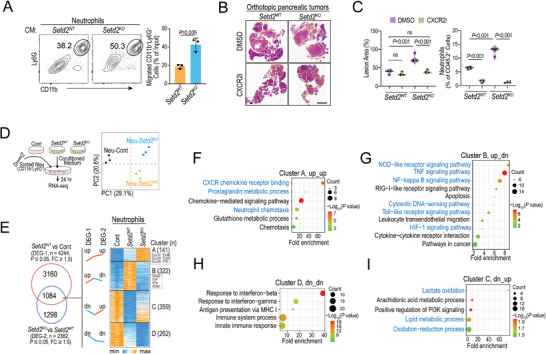
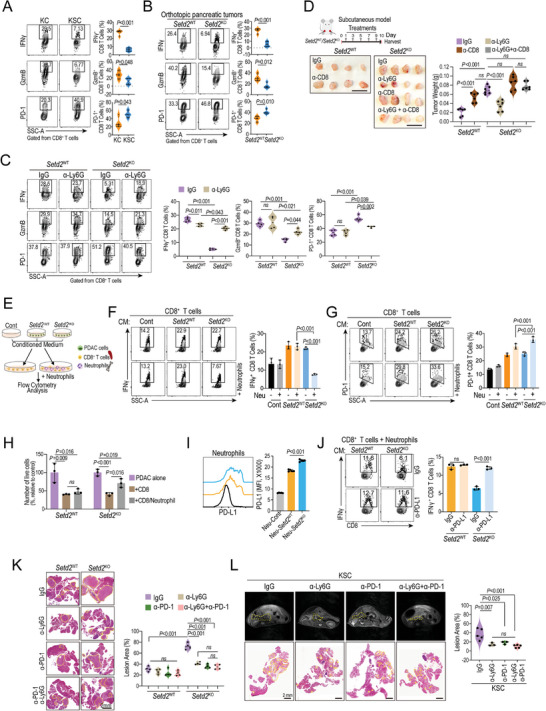
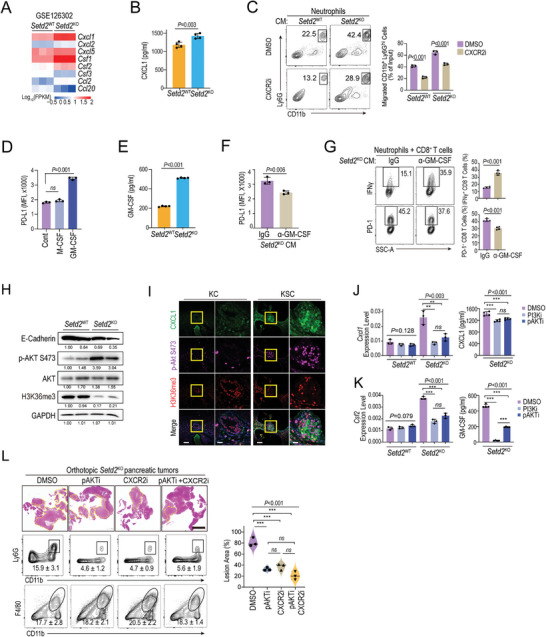
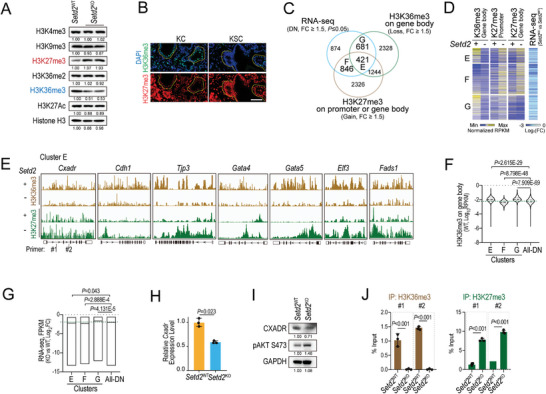
Genetic and epigenetic alterations play central roles in shaping the immunosuppressive tumor microenvironment (TME) to evade immune surveillance. The previous study shows that SETD2-H3K36me3 loss promotes KRAS-induced pancreatic tumorigenesis. However, little is known about its role in remodeling the TME and immune evasion. Here, it is shown that SETD2 deficiency can reprogram neutrophils to an immunosuppressive phenotype, thereby promoting immune escape during pancreatic tumor progression. By comprehensive profiling of the intratumoral immune cells, neutrophils are identified as the subset with the most significant changes upon Setd2 loss. Setd2-deficient pancreatic tumor cells directly enhance neutrophil recruitment and reprogramming, thereby inhibiting the cytotoxicity of CD8 T cells to foster tumor progression. Mechanistically, it is revealed that Setd2-H3K36me3 loss leads to ectopic gain of H3K27me3 to downregulate Cxadr expression, which boosts the PI3K-AKT pathway and excessive expression of CXCL1 and GM-CSF, thereby promoting neutrophil recruitment and reprogramming toward an immunosuppressive phenotype. The study provides mechanistic insights into how tumor cell-intrinsic Setd2 deficiency strengthens the immune escape during pancreatic tumorigenesis, which may offer potential therapeutic implications for pancreatic cancer patients with SETD2 deficiency.
遗传和表观遗传改变在塑造免疫抑制性肿瘤微环境(TME)以逃避免疫监视方面发挥着核心作用。先前的研究表明,SETD2-H3K36me3 的缺失促进了 KRAS 诱导的胰腺肿瘤发生。然而,其在重塑 TME 和免疫逃逸中的作用知之甚少。在这里,研究表明 SETD2 缺失可以将中性粒细胞重新编程为免疫抑制表型,从而在胰腺肿瘤进展过程中促进免疫逃逸。通过对肿瘤内免疫细胞的全面分析,鉴定出中性粒细胞是 Setd2 缺失时变化最显著的亚群。Setd2 缺失的胰腺肿瘤细胞直接增强中性粒细胞的募集和重编程,从而抑制 CD8 T 细胞的细胞毒性,促进肿瘤进展。机制上,研究揭示了 Setd2-H3K36me3 的缺失导致 H3K27me3 的异位获得,从而下调 Cxadr 的表达,从而激活 PI3K-AKT 通路,过度表达 CXCL1 和 GM-CSF,从而促进中性粒细胞募集和向免疫抑制表型的重编程。该研究为肿瘤细胞内在的 Setd2 缺失如何在胰腺肿瘤发生过程中增强免疫逃逸提供了机制见解,这可能为 SETD2 缺失的胰腺癌患者提供潜在的治疗意义。