Ghosh Subham, Chetia Dipak, Gogoi Neelutpal, Rudrapal Mithun
Deptartment of Pharmaceutical Sciences, Dibrugarh University, Dibrugarh, Assam, India.
Department of Pharmaceutical Chemistry, Rasiklal M. Dhariwal Institute of Pharmaceutical Education and Research, Chinchwad, Pune, Maharashtra, India.
BioTechnologia (Pozn). 2021 Sep 30;102(3):257-275. doi: 10.5114/bta.2021.108722. eCollection 2021.
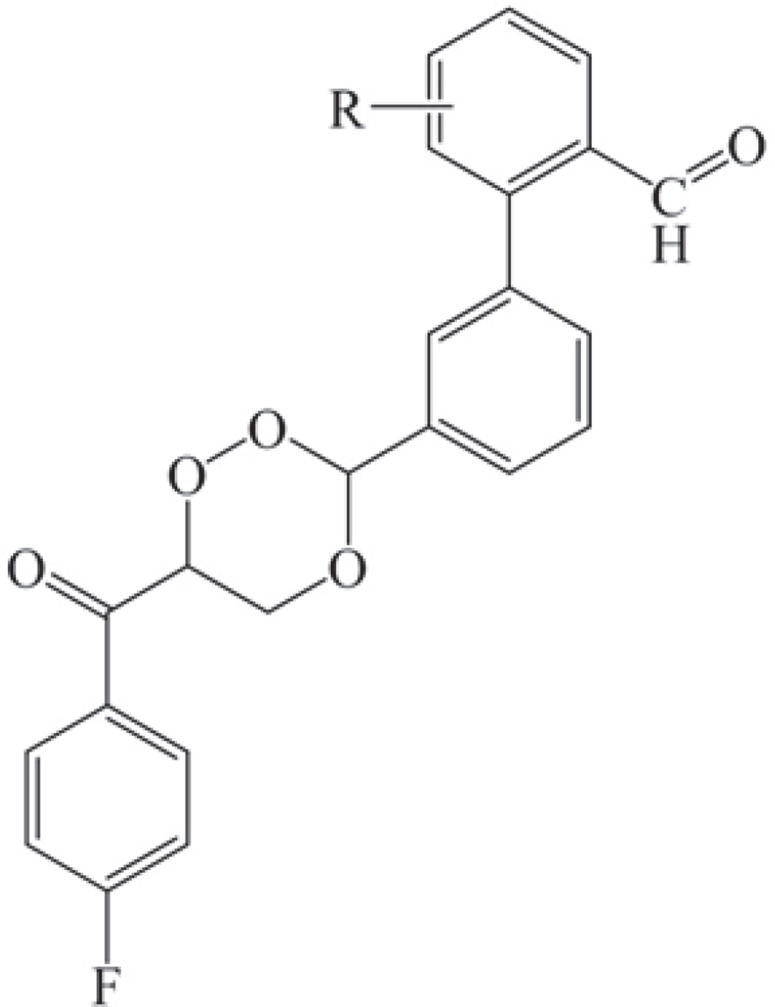
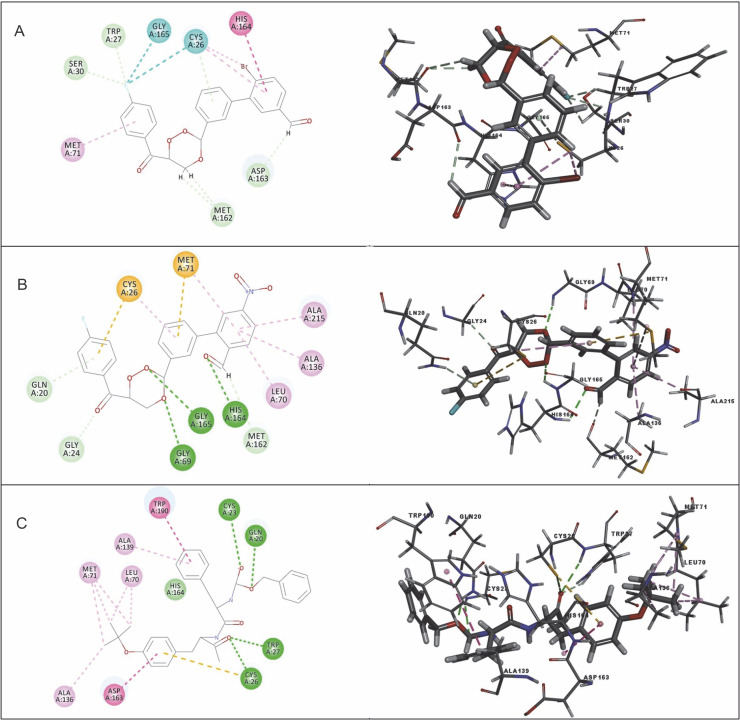
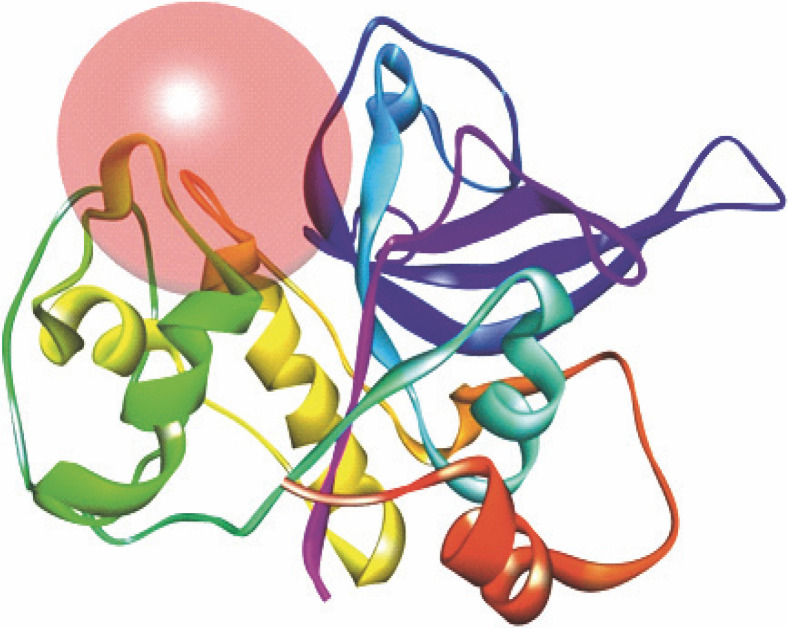
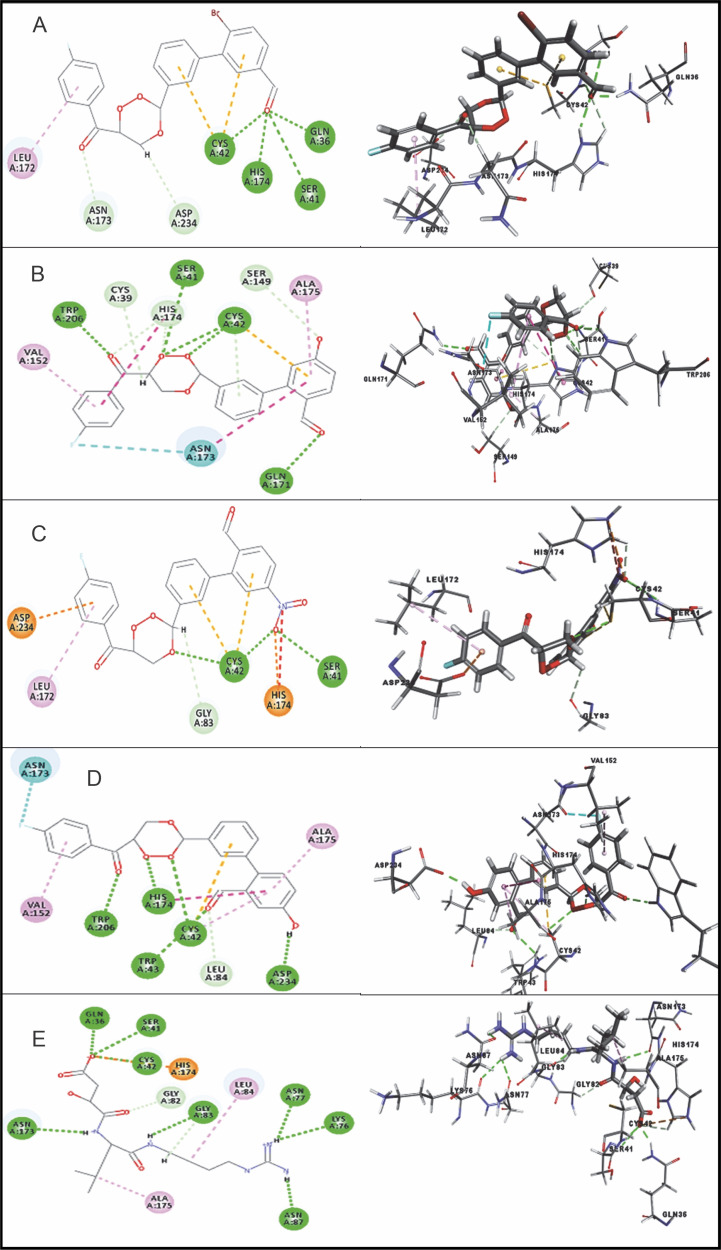
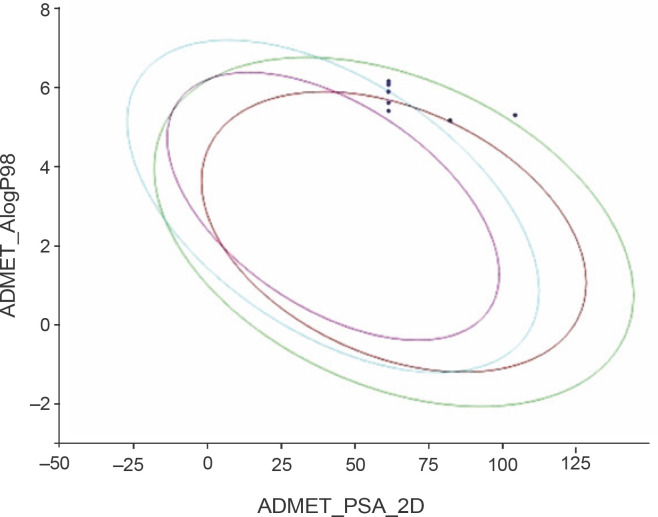
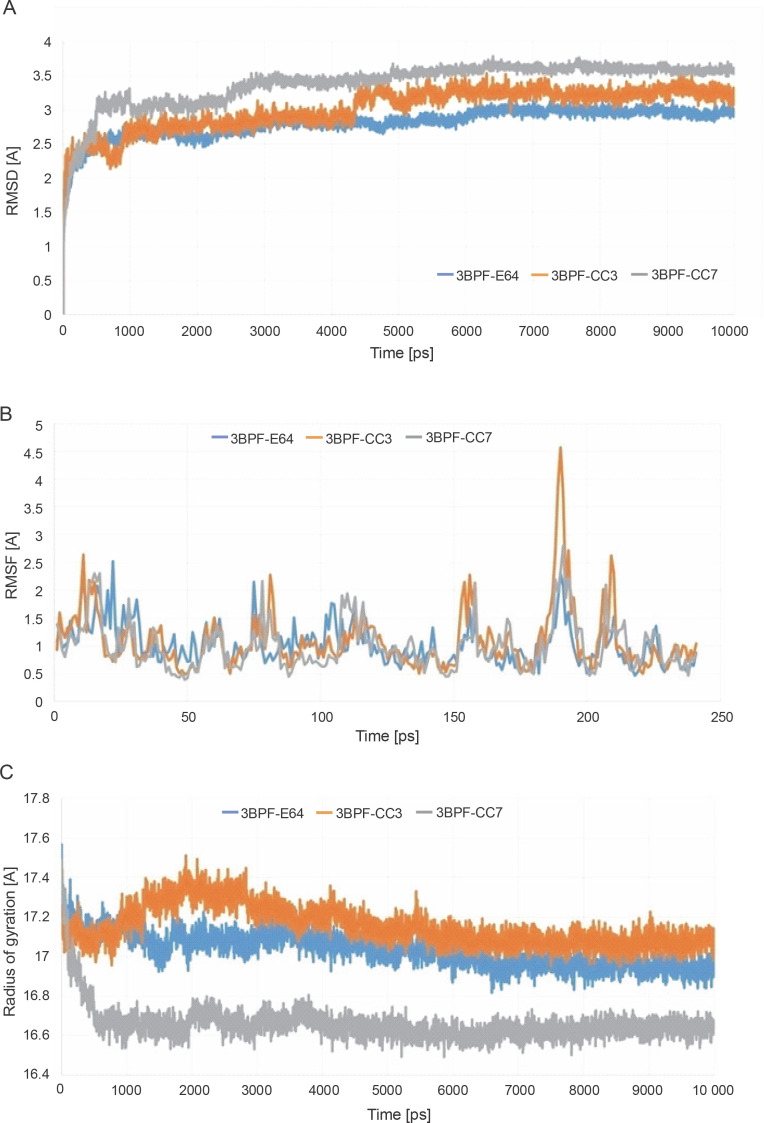
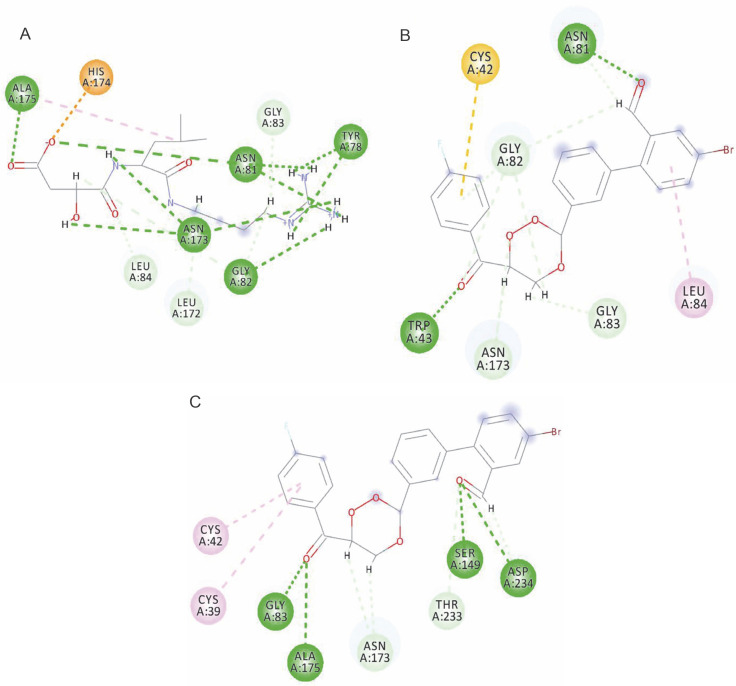
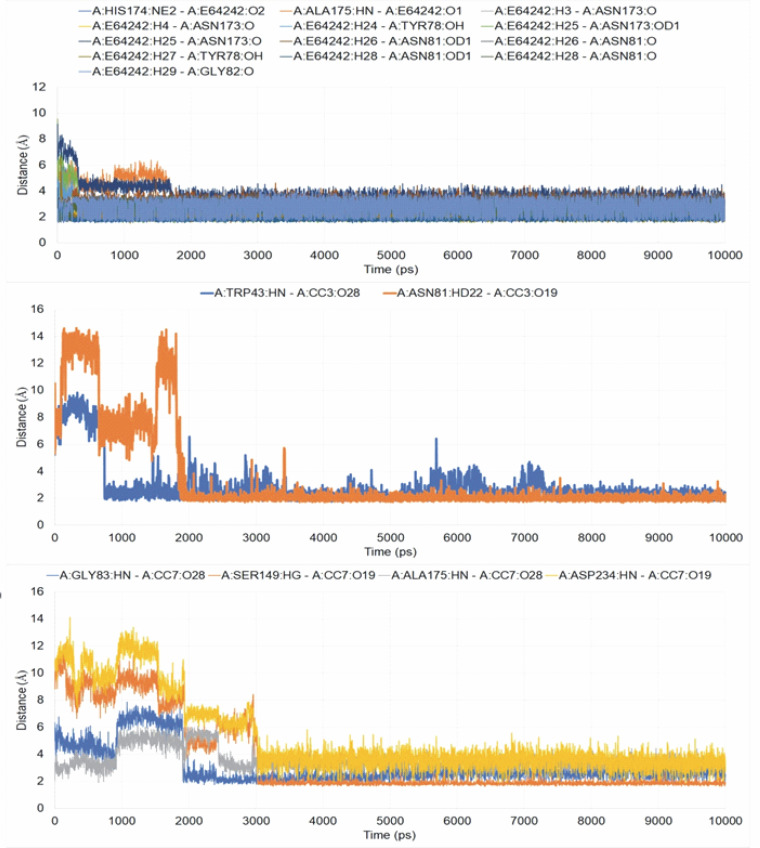
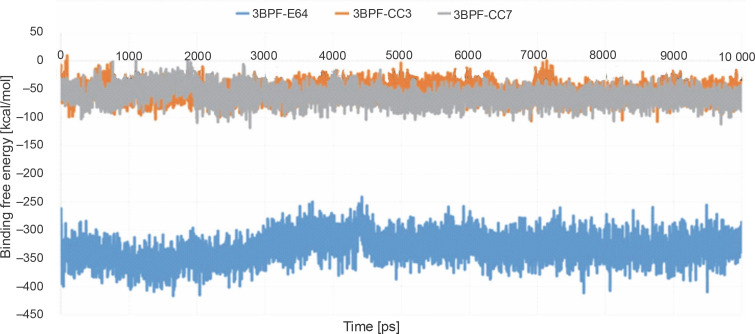
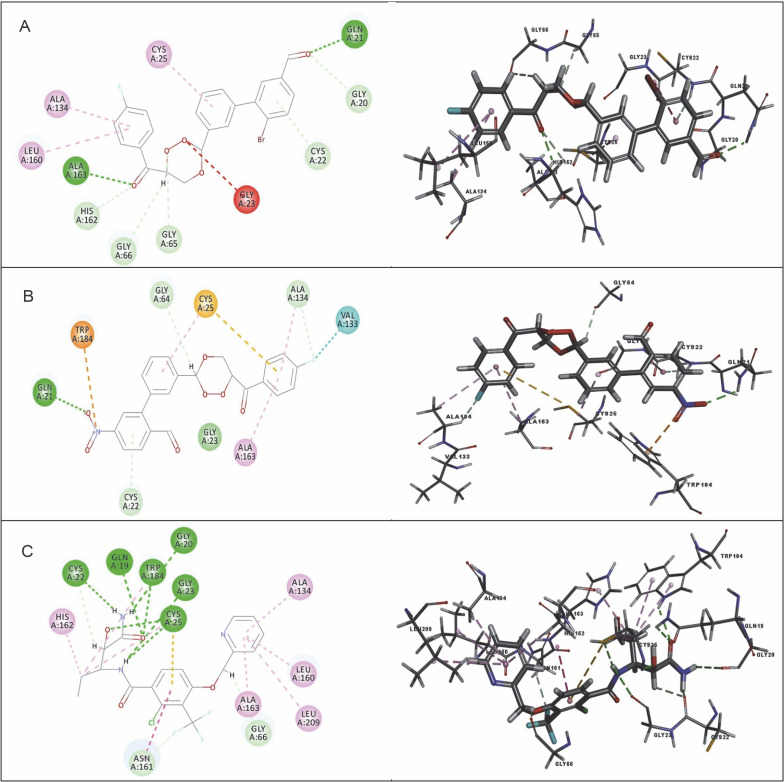
Despite significant progress made in drug discovery and development over the past few decades, malaria remains a life-threatening infectious disease across the globe. Because of the widespread emergence of drug-resistant strains of , the clinical utility of existing drug therapies including Artemisinin-based Combination Therapies (ACTs) in the treatment of malaria has been increasingly limited. It has become a serious health concern which, therefore, necessitates the development of novel drug molecules and/or alternative therapies to combat, particularly resistant . The objective of the present study was to develop 1,2,4-trioxane derivatives as novel antimalarial agents that would be effective against resistant . In our study, 15 new trioxane derivatives were designed by molecular modification of the 1,2,4-trioxane scaffold as possible antimalarial agents. Molecular modeling studies of trioxane derivatives were performed based on the CADD approach using Biovia Discovery Studio (DS) 2018 software. The protein-ligand docking study was performed against falcipain 2 (FP-2) using the simulation-based docking protocol LibDock by the flexible docking method. The assessment of drug-likeness, ADMET properties, and toxicity was also performed. Furthermore, the compounds CC3 and CC7, which showed the best binding affinity against the target FP-2, were investigated by molecular dynamics (MD) simulation studies followed by the calculation of MM-PBSA binding free energy of protein-ligand complexes using DS 2020. Results of the docking study showed that among the 15 compounds, three trioxane derivatives were found to possess promising binding affinity with LibDock scores ranging from 117.16 to 116.90. Drug-likeness, ADMET, and toxicity properties were found to be satisfactory for all the compounds. Among the 15 compounds, two compounds, namely CC3 and CC7, showed the highest binding affinity against FP-2 with LibDock score of 117.166 and 117.200, respectively. The Libdock score of the co-crystal inhibitor was 114.474. MD studies along with MM-PBSA calculations of binding energies further confirmed the antimalarial potential of the compounds CC3 and CC7, with the formation of well-defined and stable receptor-ligand interactions against the FP-2 enzyme. Additionally, the selectivity of trioxane hits identified as potential inhibitors of cysteine protease FP-2 was determined on human cysteine proteases such as cathepsins (Cat K and Cat L), which are host homologous. Finally, it was concluded that the newly designed 1,2,4-trioxane derivatives can be further studied for and antimalarial activities for their possible development as potent antimalarial agents effective against resistant .
尽管在过去几十年里药物研发取得了重大进展,但疟疾在全球范围内仍然是一种危及生命的传染病。由于疟原虫耐药菌株的广泛出现,包括青蒿素联合疗法(ACTs)在内的现有药物疗法在疟疾治疗中的临床效用越来越有限。这已成为一个严重的健康问题,因此,有必要开发新型药物分子和/或替代疗法来对抗疟疾,特别是耐药疟原虫。本研究的目的是开发1,2,4-三恶烷衍生物作为新型抗疟药物,以有效对抗耐药疟原虫。在我们的研究中,通过对1,2,4-三恶烷骨架进行分子修饰,设计了15种新的三恶烷衍生物作为潜在的抗疟药物。使用Biovia Discovery Studio(DS)2018软件,基于计算机辅助药物设计(CADD)方法对三恶烷衍生物进行了分子建模研究。通过灵活对接方法,使用基于模拟的对接协议LibDock,针对恶性疟原虫木瓜蛋白酶2(FP-2)进行了蛋白质-配体对接研究。还对药物相似性、ADMET性质和毒性进行了评估。此外,对与目标FP-2表现出最佳结合亲和力的化合物CC3和CC7进行了分子动力学(MD)模拟研究,随后使用DS 2020计算了蛋白质-配体复合物的MM-PBSA结合自由能。对接研究结果表明,在这15种化合物中,发现三种三恶烷衍生物具有良好的结合亲和力,LibDock分数在117.16至116.90之间。发现所有化合物的药物相似性、ADMET和毒性性质均令人满意。在这15种化合物中,两种化合物CC3和CC7对FP-2表现出最高的结合亲和力,LibDock分数分别为117.166和117.200。共晶体抑制剂的Libdock分数为114.474。MD研究以及结合能的MM-PBSA计算进一步证实了化合物CC3和CC7的抗疟潜力,它们与FP-2酶形成了明确且稳定的受体-配体相互作用。此外,在人半胱氨酸蛋白酶如组织蛋白酶(组织蛋白酶K和组织蛋白酶L)上测定了被鉴定为半胱氨酸蛋白酶FP-2潜在抑制剂的三恶烷命中物的选择性,这些组织蛋白酶是宿主同源的。最后得出结论,新设计的1,2,4-三恶烷衍生物可进一步研究其对耐药疟原虫的体内和体外抗疟活性,以开发成为有效的新型抗疟药物。