Medical Genetics Section, University Hospital Consortium Corporation Polyclinics of Bari, 70124 Bari, Italy.
U.O.C. Genetica Medica e di Laboratorio, Ospedale Antonio Cardarelli, 80131 Napoli, Italy.
Genes (Basel). 2023 Jan 7;14(1):165. doi: 10.3390/genes14010165.
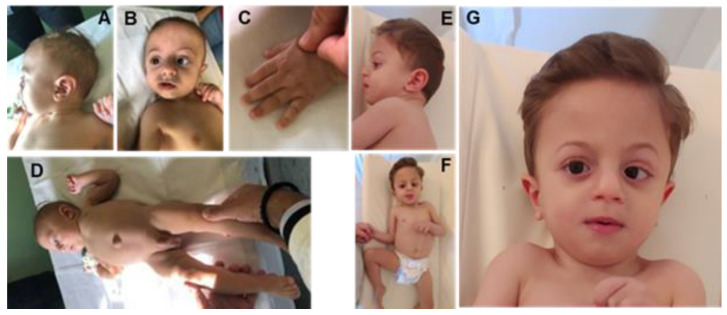
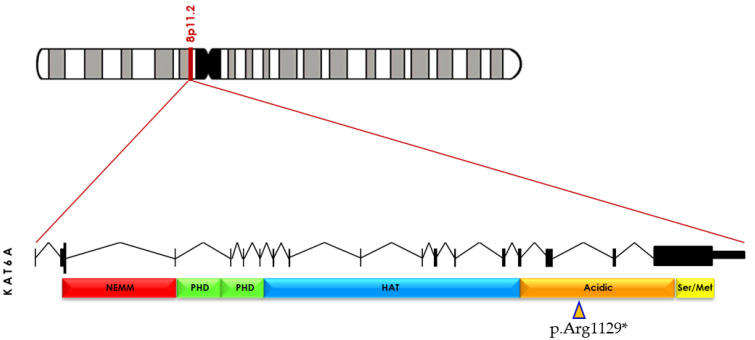
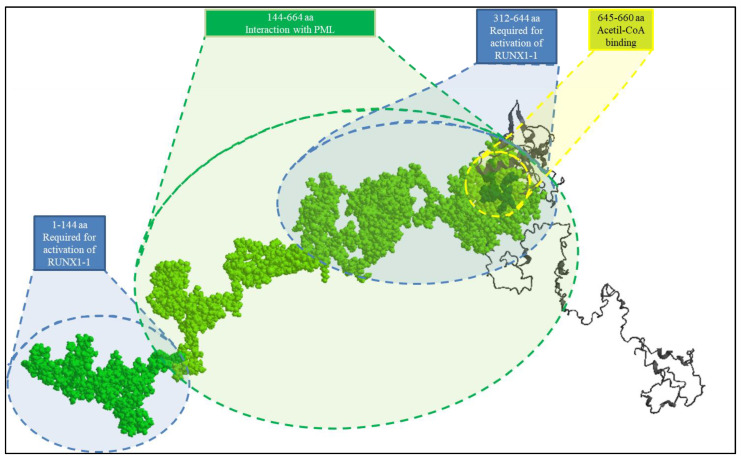
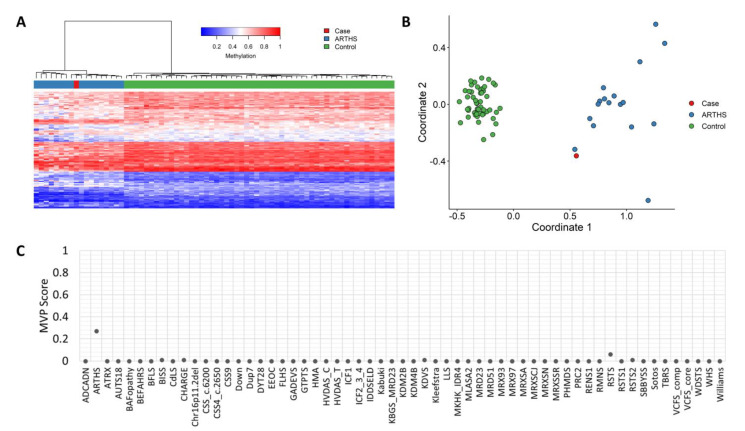
Pathogenic variants in genes are involved in histone acetylation and deacetylation resulting in congenital anomalies, with most patients displaying a neurodevelopmental disorder and dysmorphism. Arboleda-Tham syndrome caused by pathogenic variants in KAT6A (Lysine Acetyltransferase 6A; OMIM 601408) has been recently described as a new neurodevelopmental disorder. Herein, we describe a patient characterized by complex phenotype subsequently diagnosed using the clinical exome sequencing (CES) with Arboleda-Tham syndrome (ARTHS; OMIM 616268). The analysis revealed the presence of de novo pathogenic variant in KAT6A gene, a nucleotide c.3385C>T substitution that introduces a premature termination codon (p.Arg1129*). The need for straight multidisciplinary collaboration and accurate clinical description findings (bowel obstruction/megacolon/intestinal malrotation) was emphasized, together with the utility of CES in establishing an etiological basis in clinical and genetical heterogeneous conditions. Therefore, considering the phenotypic characteristics, the condition’s rarity and the reviewed literature, we propose additional diagnostic criteria that could help in the development of future clinical diagnostic guidelines. This was possible thanks to objective examinations performed during the long follow-up period, which permitted scrupulous registration of phenotypic changes over time to further assess this rare disorder. Finally, given that different genetic syndromes are associated with distinct genomic DNA methylation patterns used for diagnostic testing and/or as biomarker of disease, a specific episignature for ARTHS has been identified.
基因中的致病变异参与组蛋白乙酰化和去乙酰化,导致先天性异常,大多数患者表现出神经发育障碍和发育异常。由 KAT6A(赖氨酸乙酰转移酶 6A;OMIM 601408)中的致病变异引起的 Arboleda-Tham 综合征最近被描述为一种新的神经发育障碍。在此,我们描述了一位患者,其具有复杂的表型,随后使用临床外显子组测序(CES)诊断为 Arboleda-Tham 综合征(ARTHS;OMIM 616268)。分析显示 KAT6A 基因存在新生致病变异,核苷酸 c.3385C>T 取代导致提前终止密码子(p.Arg1129*)。强调了需要直接的多学科合作和准确的临床描述发现(肠梗阻/巨结肠/肠旋转不良),以及 CES 在建立临床和遗传异质性条件的病因基础方面的实用性。因此,考虑到表型特征、该病症的罕见性和已审查的文献,我们提出了其他诊断标准,这可能有助于未来临床诊断指南的制定。这要归功于在长期随访期间进行的客观检查,这些检查能够严格记录随时间推移的表型变化,从而进一步评估这种罕见疾病。最后,鉴于不同的遗传综合征与用于诊断测试和/或作为疾病生物标志物的不同基因组 DNA 甲基化模式相关,已经确定了 ARTHS 的特定表观遗传签名。