Kazmirczak Felipe, Vogel Neal T, Prisco Sasha Z, Patterson Michael T, Annis Jeffrey, Moon Ryan T, Hartweck Lynn M, Mendelson Jenna B, Kim Minwoo, Mancipe Natalia Calixto, Markowski Todd, Higgins LeAnn, Guerrero Candace, Kremer Ben, Blake Madelyn L, Rhodes Christopher J, Williams Jesse W, Brittain Evan L, Prins Kurt W
Minneapolis Heart Institute, Minneapolis, MN.
Lillehei Heart Institute, Cardiovascular Division, Department of Medicine, University of Minnesota, Minneapolis, MN.
bioRxiv. 2024 Sep 23:2023.01.19.524721. doi: 10.1101/2023.01.19.524721.
Mitochondrial dysfunction, characterized by impaired lipid metabolism and heightened reactive oxygen species (ROS) generation, results in lipid peroxidation and ferroptosis. Ferroptosis is an inflammatory mode of cell death that promotes complement activation and macrophage recruitment. In pulmonary arterial hypertension (PAH), pulmonary arterial endothelial cells (PAEC) exhibit cellular phenotypes that promote ferroptosis. Moreover, there is ectopic complement deposition and inflammatory macrophage accumulation in the pulmonary vasculature. However, the effects of ferroptosis inhibition on these pathogenic mechanisms and the cellular landscape of the pulmonary vasculature are incompletely defined.
Multi-omics and physiological analyses evaluated how ferroptosis inhibition modulated preclinical PAH. The impact of AAV1-mediated expression of the pro-ferroptotic protein ACSL4 on PAH was determined, and a genetic association study in humans further probed the relationship between ferroptosis and pulmonary hypertension (PH).
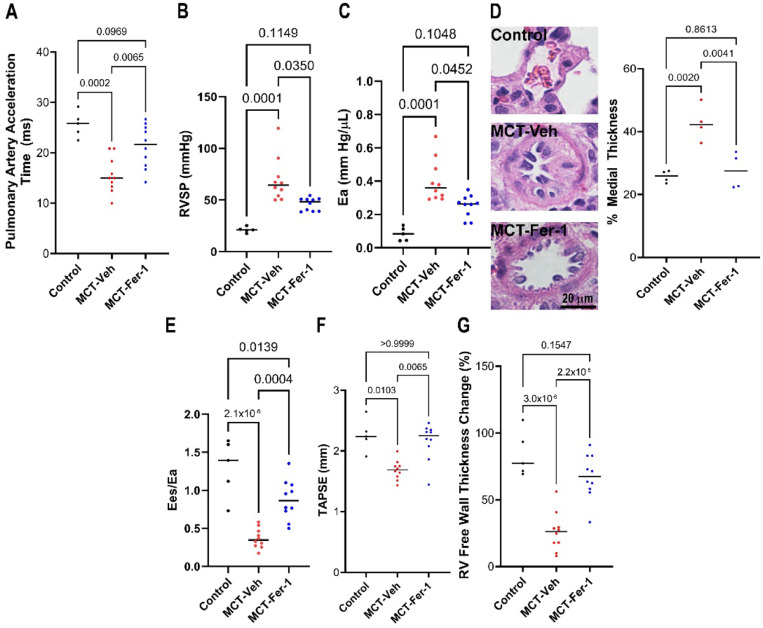
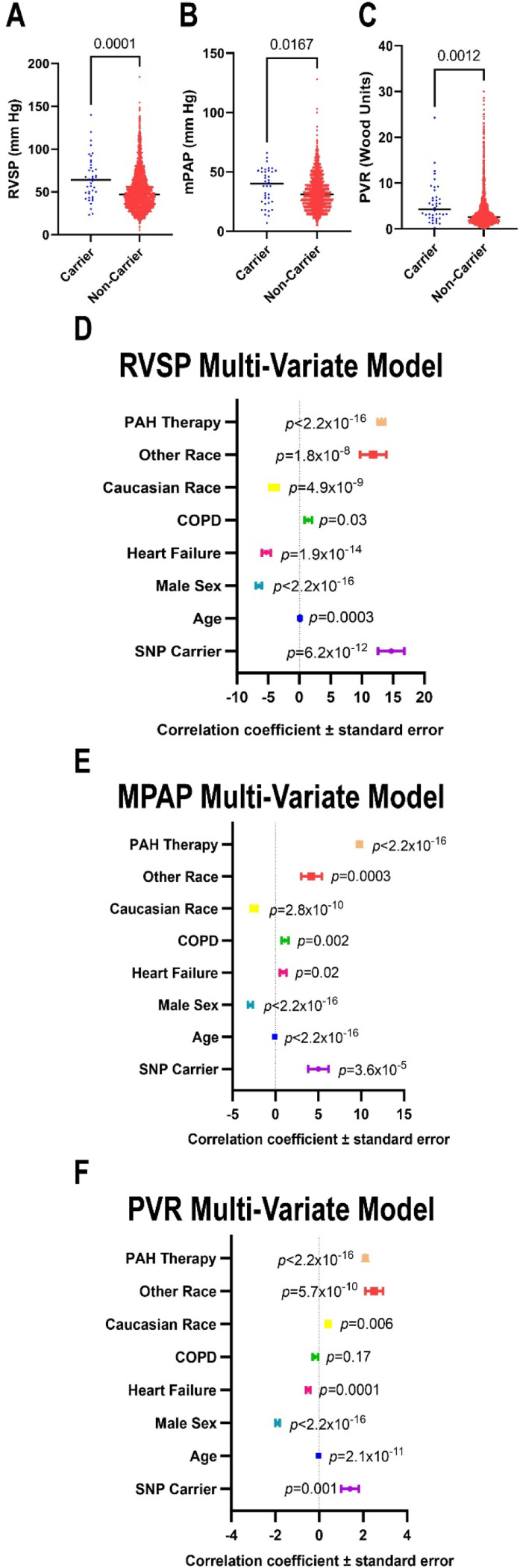
Ferrostatin-1, a small-molecule ferroptosis inhibitor, mitigated PAH severity in monocrotaline rats. RNA-seq and proteomics analyses demonstrated ferroptosis was associated with PAH severity. RNA-seq, proteomics, and confocal microscopy revealed complement activation and pro-inflammatory cytokines/chemokines were suppressed by ferrostatin-1. Additionally, ferrostatin-1 combatted changes in endothelial, smooth muscle, and interstitial macrophage abundance and gene activation patterns as revealed by deconvolution RNA-seq. Ferroptotic PAEC damage associated molecular patterns restructured the transcriptomic signature, mitochondrial morphology, and promoted proliferation of pulmonary artery smooth muscle cells, and created a pro-inflammatory phenotype in monocytes . AAV1- induced an inflammatory PAH phenotype in rats. Finally, single-nucleotide polymorphisms in six ferroptosis genes identified a potential link between ferroptosis and PH severity in the Vanderbilt BioVU repository.
Ferroptosis promotes PAH through metabolic and inflammatory mechanisms in the pulmonary vasculature.
线粒体功能障碍以脂质代谢受损和活性氧(ROS)生成增加为特征,会导致脂质过氧化和铁死亡。铁死亡是一种促进补体激活和巨噬细胞募集的细胞死亡炎症模式。在肺动脉高压(PAH)中,肺动脉内皮细胞(PAEC)表现出促进铁死亡的细胞表型。此外,肺血管中存在异位补体沉积和炎性巨噬细胞积聚。然而,铁死亡抑制对这些致病机制和肺血管细胞格局的影响尚未完全明确。
多组学和生理学分析评估了铁死亡抑制如何调节临床前PAH。确定了腺相关病毒1(AAV1)介导的促铁死亡蛋白ACSL4表达对PAH的影响,并且在人类中进行的一项基因关联研究进一步探究了铁死亡与肺动脉高压(PH)之间的关系。
小分子铁死亡抑制剂铁抑素-1减轻了野百合碱诱导的大鼠PAH严重程度。RNA测序(RNA-seq)和蛋白质组学分析表明,铁死亡与PAH严重程度相关。RNA-seq、蛋白质组学和共聚焦显微镜显示,铁抑素-1抑制了补体激活以及促炎细胞因子/趋化因子。此外,如去卷积RNA-seq所揭示的,铁抑素-1对抗了内皮细胞、平滑肌细胞和间质巨噬细胞丰度以及基因激活模式的变化。铁死亡的PAEC损伤相关分子模式重塑了转录组特征、线粒体形态,并促进了肺动脉平滑肌细胞的增殖,还在单核细胞中产生了促炎表型。AAV1在大鼠中诱导了炎性PAH表型。最后,在范德比尔特生物样本库中,六个铁死亡基因中的单核苷酸多态性确定了铁死亡与PH严重程度之间的潜在联系。
铁死亡通过肺血管中的代谢和炎症机制促进PAH。