Zhang Zhenjie, Ma Mingsheng, Zhang Weimin, Zhou Yu, Yao Fengxia, Zhu Lisi, Wei Min, Qiu Zhengqing
Department of Pediatrics, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China.
Department of Genetics Laboratory, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China.
Front Genet. 2023 Jan 13;14:1103620. doi: 10.3389/fgene.2023.1103620. eCollection 2023.
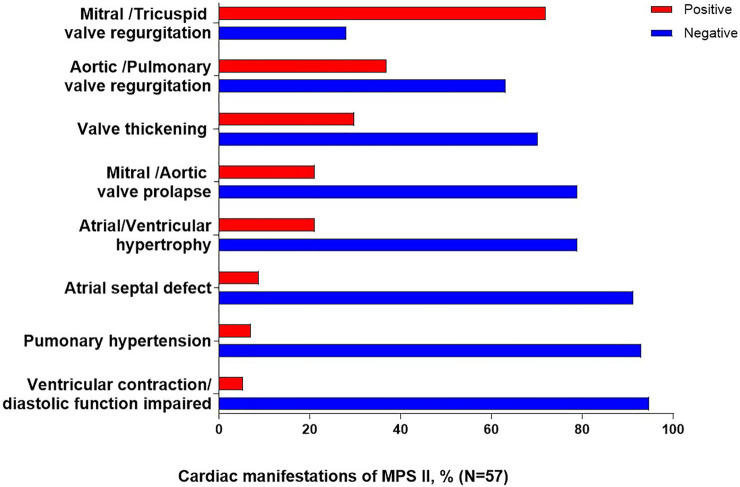
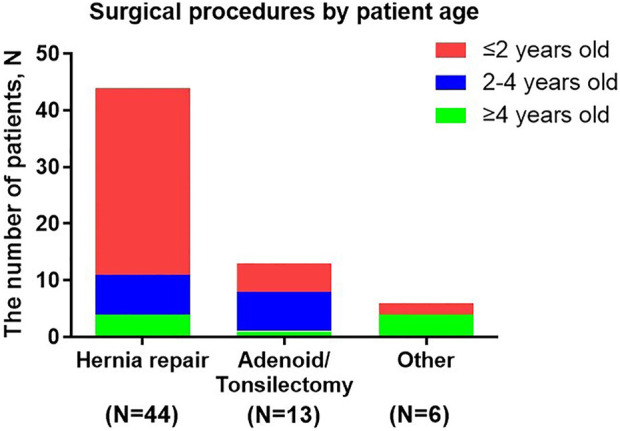
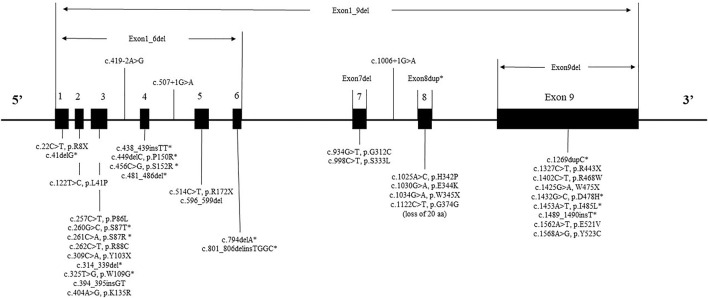
Mucopolysaccharidosis Type II (MPS II) is a rare, progressive and ultimately fatal X-linked lysosomal storage disorder caused by mutations in the iduronate-2-sulfatase (IDS) gene. This report conducted a retrospective analysis to investigate the clinical characteristics, genotypes and management strategies in a large cohort of Chinese patients with MPS II. In this study, we explored 130 Chinese patients with MPS II between September 2008 and April 2022. Clinical manifestations, auxiliary examination, IDS pathogenic gene variants and IDS enzyme activity, surgical history were analysed in the study. A total of 130 patients were enrolled and the mean age at diagnosis was 5 years old. This study found the most common symptoms in our patients were claw-like hands, followed by coarse facial features, birthmarks (Mongolian spot), delayed development, inguinal or umbilical hernia. The most commonly cardiac manifestations were valve abnormalities, which were mitral/tricuspid valve regurgitation (71.9%) and aortic/pulmonary valve regurgitation (36.8%). We had found 43 different IDS pathogenic gene variants in 55 patients, included 16 novel variants. The variants were concentrated in exon 9 (20% = 11/55), exon 3 (20% = 11/55) and exon 8 (15% = 8/55). A total of 50 patients (38.5%) underwent surgical treatment, receiving a total of 63 surgeries. The average age of first surgery was 2.6 years, and the majority of surgery (85.7%, 54/63) was operated before 4 years old. The most common and earliest surgery was hernia repair. Three patients were died of respiratory failure. This study provided additional information on the clinical, cardiac ultrasound and surgical procedure in MPS II patients. Our study expanded the genotype spectrum of MPS II. Based on these data, characterization of MPS II patients group could be used to early diagnosis and treatment of the disease.
II型黏多糖贮积症(MPS II)是一种罕见的、进行性且最终致命的X连锁溶酶体贮积症,由艾杜糖醛酸-2-硫酸酯酶(IDS)基因突变引起。本报告进行了一项回顾性分析,以调查一大群中国MPS II患者的临床特征、基因型和管理策略。在本研究中,我们对2008年9月至2022年4月期间的130例中国MPS II患者进行了探索。对研究中的临床表现、辅助检查、IDS致病基因变异和IDS酶活性、手术史进行了分析。共纳入130例患者,诊断时的平均年龄为5岁。本研究发现,我们患者中最常见的症状是爪状手,其次是面部粗糙、胎记(蒙古斑)、发育迟缓、腹股沟或脐疝。最常见的心脏表现是瓣膜异常,即二尖瓣/三尖瓣反流(71.9%)和主动脉瓣/肺动脉瓣反流(36.8%)。我们在55例患者中发现了43种不同的IDS致病基因变异,其中包括16种新变异。这些变异集中在外显子9(20% = 11/55)、外显子3(20% = 11/55)和外显子8(15% = 8/55)。共有50例患者(38.5%)接受了手术治疗,共进行了63次手术。首次手术的平均年龄为2.6岁,大多数手术(85.7%,54/63)在4岁之前进行。最常见和最早进行的手术是疝气修补术。3例患者死于呼吸衰竭。本研究提供了有关MPS II患者临床、心脏超声和手术过程的更多信息。我们的研究扩展了MPS II的基因型谱。基于这些数据,MPS II患者群体的特征可用于该疾病的早期诊断和治疗。