Loxo@Lilly, Louisville, CO.
Loxo@Lilly, South San Francisco, CA.
Blood. 2023 Jul 6;142(1):62-72. doi: 10.1182/blood.2022018674.
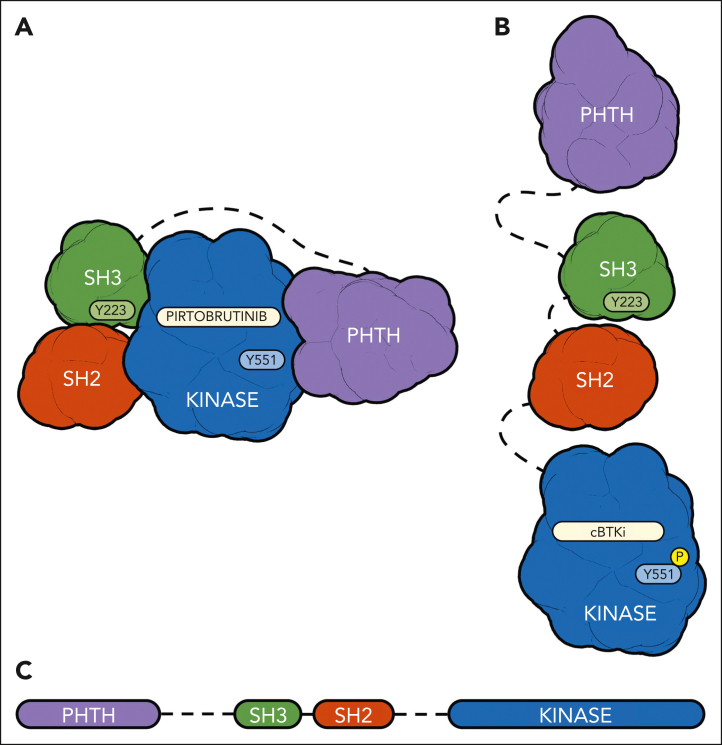
Bruton tyrosine kinase (BTK), a nonreceptor tyrosine kinase, is a major therapeutic target for B-cell-driven malignancies. However, approved covalent BTK inhibitors (cBTKis) are associated with treatment limitations because of off-target side effects, suboptimal oral pharmacology, and development of resistance mutations (eg, C481) that prevent inhibitor binding. Here, we describe the preclinical profile of pirtobrutinib, a potent, highly selective, noncovalent (reversible) BTK inhibitor. Pirtobrutinib binds BTK with an extensive network of interactions to BTK and water molecules in the adenosine triphosphate binding region and shows no direct interaction with C481. Consequently, pirtobrutinib inhibits both BTK and BTK C481 substitution mutants in enzymatic and cell-based assays with similar potencies. In differential scanning fluorimetry studies, BTK bound to pirtobrutinib exhibited a higher melting temperature than cBTKi-bound BTK. Pirtobrutinib, but not cBTKis, prevented Y551 phosphorylation in the activation loop. These data suggest that pirtobrutinib uniquely stabilizes BTK in a closed, inactive conformation. Pirtobrutinib inhibits BTK signaling and cell proliferation in multiple B-cell lymphoma cell lines, and significantly inhibits tumor growth in human lymphoma xenografts in vivo. Enzymatic profiling showed that pirtobrutinib was highly selective for BTK in >98% of the human kinome, and in follow-up cellular studies pirtobrutinib retained >100-fold selectivity over other tested kinases. Collectively, these findings suggest that pirtobrutinib represents a novel BTK inhibitor with improved selectivity and unique pharmacologic, biophysical, and structural attributes with the potential to treat B-cell-driven cancers with improved precision and tolerability. Pirtobrutinib is being tested in phase 3 clinical studies for a variety of B-cell malignancies.
布鲁顿酪氨酸激酶(BTK)是一种非受体酪氨酸激酶,是驱动 B 细胞恶性肿瘤的主要治疗靶点。然而,已批准的共价 BTK 抑制剂(cBTKis)由于脱靶副作用、不理想的口服药代动力学和耐药突变(如 C481)的发展而导致抑制剂结合受阻,存在治疗局限性。在这里,我们描述了强效、高度选择性的非共价(可逆)BTK 抑制剂 pirtobrutinib 的临床前特征。Pirtobrutinib 通过广泛的相互作用网络与 BTK 和三磷酸腺苷结合区域的水分子结合,与 C481 没有直接相互作用。因此,Pirtobrutinib 在酶和基于细胞的测定中以相似的效力抑制 BTK 和 BTK C481 取代突变体。在差示扫描荧光法研究中,与 pirtobrutinib 结合的 BTK 比与 cBTKi 结合的 BTK 的熔点更高。Pirtobrutinib,但不是 cBTKis,可防止激活环中的 Y551 磷酸化。这些数据表明 pirtobrutinib 独特地将 BTK 稳定在闭合的、非活性构象中。Pirtobrutinib 抑制多种 B 细胞淋巴瘤细胞系中的 BTK 信号传导和细胞增殖,并显著抑制体内人淋巴瘤异种移植中的肿瘤生长。酶谱分析表明,在 >98%的人类激酶组中,Pirtobrutinib 对 BTK 具有高度选择性,在后续的细胞研究中,Pirtobrutinib 对其他测试激酶保持 >100 倍的选择性。总的来说,这些发现表明 pirtobrutinib 代表了一种新型 BTK 抑制剂,具有改善的选择性和独特的药理学、生物物理和结构特性,有可能以更高的精度和耐受性治疗 B 细胞驱动的癌症。Pirtobrutinib 正在多种 B 细胞恶性肿瘤的 3 期临床试验中进行测试。