Cafournet Cérane, Zanin Sofia, Guimier Anne, Hully Marie, Assouline Zahra, Barcia Giulia, de Lonlay Pascale, Steffann Julie, Munnich Arnold, Bonnefont Jean-Paul, Rötig Agnès, Ruzzenente Benedetta, Metodiev Metodi D
Laboratory for Genetics of Mitochondrial Disorders, Imagine Institute, Université Paris Cité, INSERM U1163, 75015 Paris, France.
Laboratory of Embryology and Genetics of Human Malformations, Imagine Institute, Université Paris Cité, INSERM U1163, 75015 Paris, France.
Life (Basel). 2023 Feb 4;13(2):445. doi: 10.3390/life13020445.
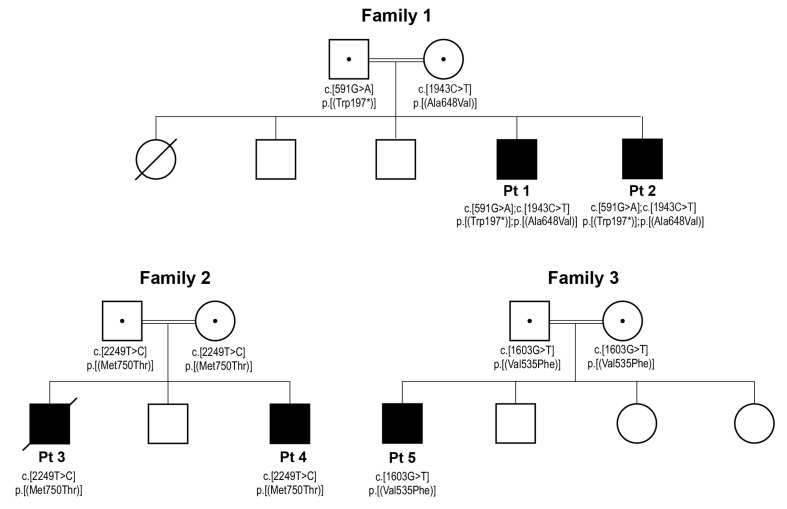
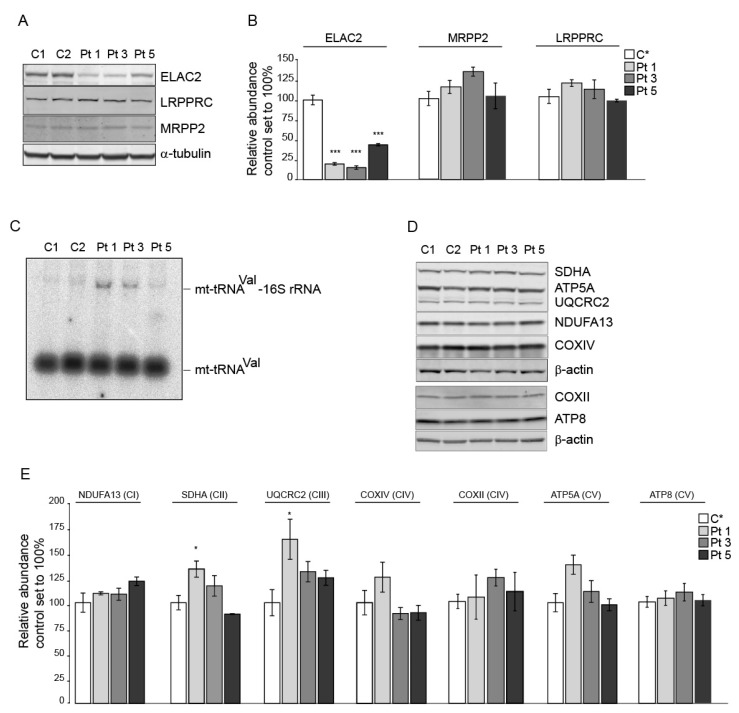
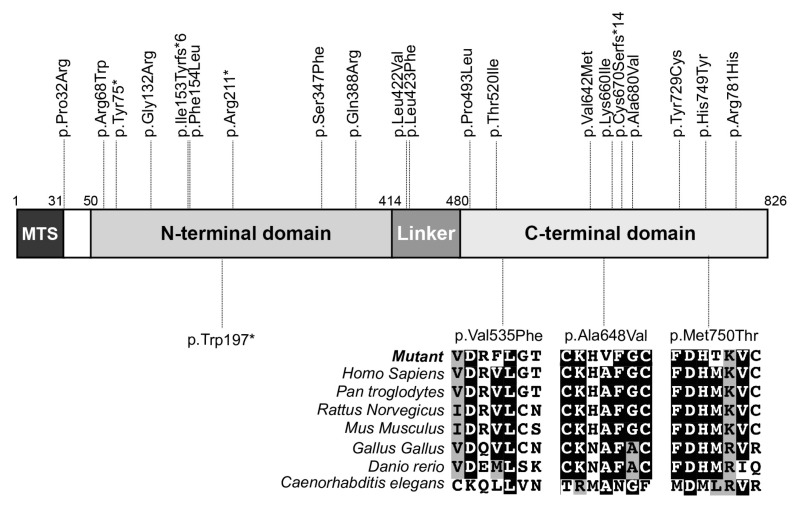
Transcription of mitochondrial DNA generates long polycistronic precursors whose nucleolytic cleavage yields the individual mtDNA-encoded transcripts. In most cases, this cleavage occurs at the 5'- and 3'-ends of tRNA sequences by the concerted action of RNAseP and RNaseZ/ELAC2 endonucleases, respectively. Variants in the gene have been predominantly linked to severe to mild cardiomyopathy that, in its milder forms, is accompanied by variably severe neurological presentations. Here, we report five patients from three unrelated families. Four of the patients presented mild to moderate cardiomyopathy and one died at 1 year of age, one patient had no evidence of cardiomyopathy. The patients had variable neurological presentations that included intellectual disability, ataxia, refractory epilepsy, neuropathy and deafness. All patients carried previously unreported missense and nonsense variants. Enzymatic analyses showed multiple OXPHOS deficiencies in biopsies from two patients, whereas immunoblot analyses revealed a decreased abundance of ELAC2 in fibroblasts from three patients. Northern blot analysis revealed an accumulation of unprocessed mt-tRNA-precursor consistent with the role of ELAC2 in transcript processing. Our study expands the genetic spectrum of -linked disease and suggests that cardiomyopathy is not an invariably present clinical hallmark of this pathology.
线粒体DNA转录产生长的多顺反子前体,其核酸裂解产生单个线粒体DNA编码的转录本。在大多数情况下,这种裂解分别通过RNAseP和RNaseZ/ELAC2核酸内切酶的协同作用,发生在tRNA序列的5'端和3'端。该基因的变异主要与严重至轻度的心肌病相关,在较轻的形式中,伴有不同程度的严重神经学表现。在此,我们报告了来自三个无关家庭的五名患者。其中四名患者表现为轻度至中度心肌病,一名患者在1岁时死亡,一名患者没有心肌病证据。这些患者有不同的神经学表现,包括智力残疾、共济失调、难治性癫痫、神经病变和耳聋。所有患者都携带了以前未报告的错义突变和无义突变。酶分析显示两名患者活检中有多种氧化磷酸化缺陷,而免疫印迹分析显示三名患者成纤维细胞中ELAC2丰度降低。Northern印迹分析显示未加工的线粒体tRNA前体积累,这与ELAC2在转录本加工中的作用一致。我们的研究扩展了该相关疾病的遗传谱,并表明心肌病并非这种病理的必然临床特征。