Department of Genetics, University of Pennsylvania, Philadelphia, PA, USA.
Division of Human Genetics, Children's Hospital of Philadelphia, Philadelphia, PA, USA.
Nat Commun. 2023 Mar 3;14(1):1230. doi: 10.1038/s41467-023-36585-y.
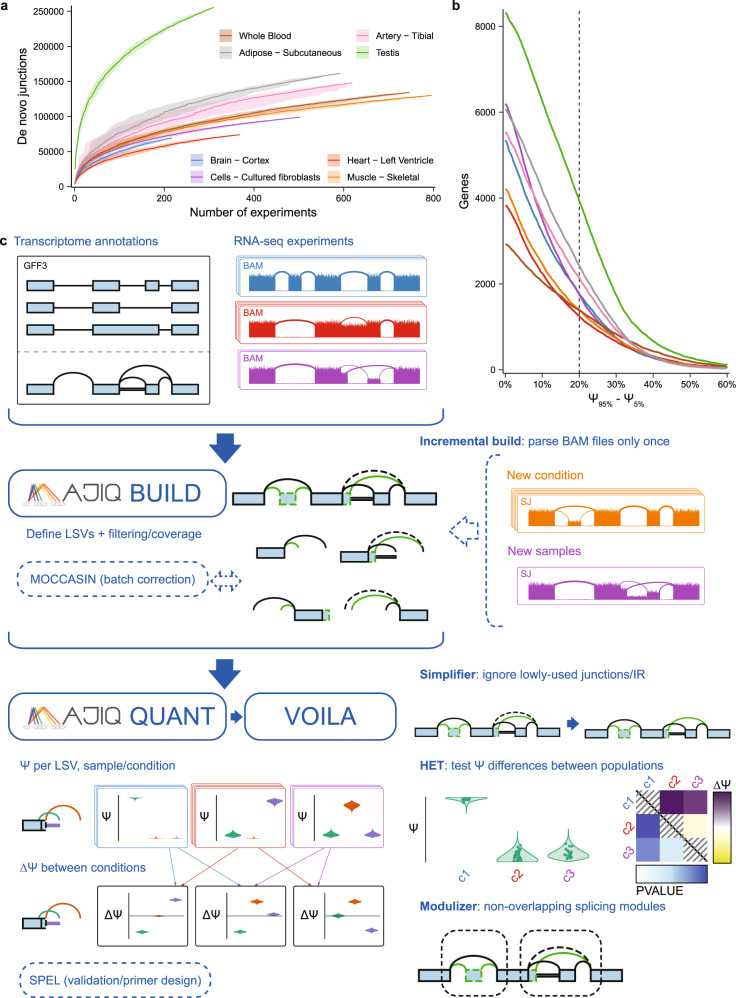
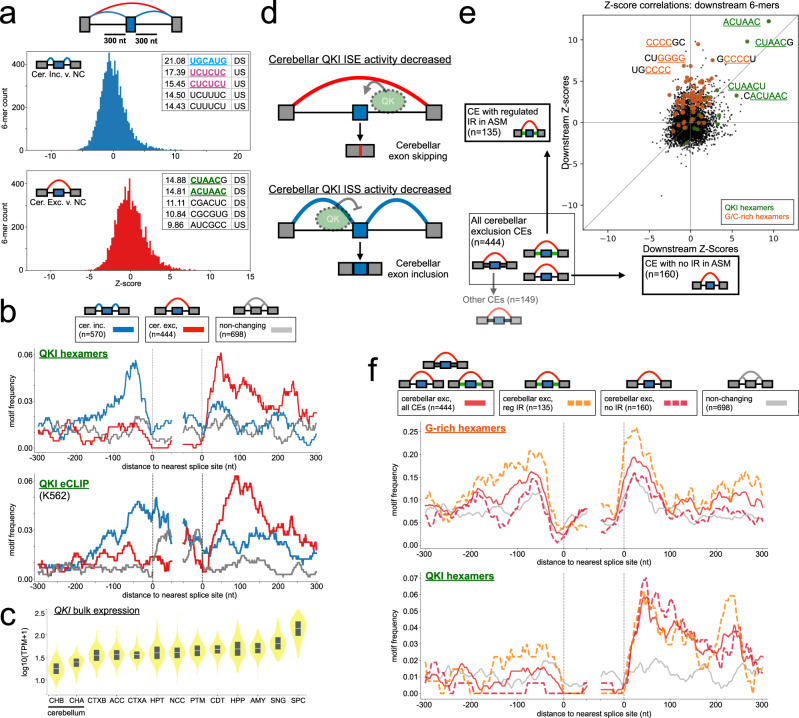
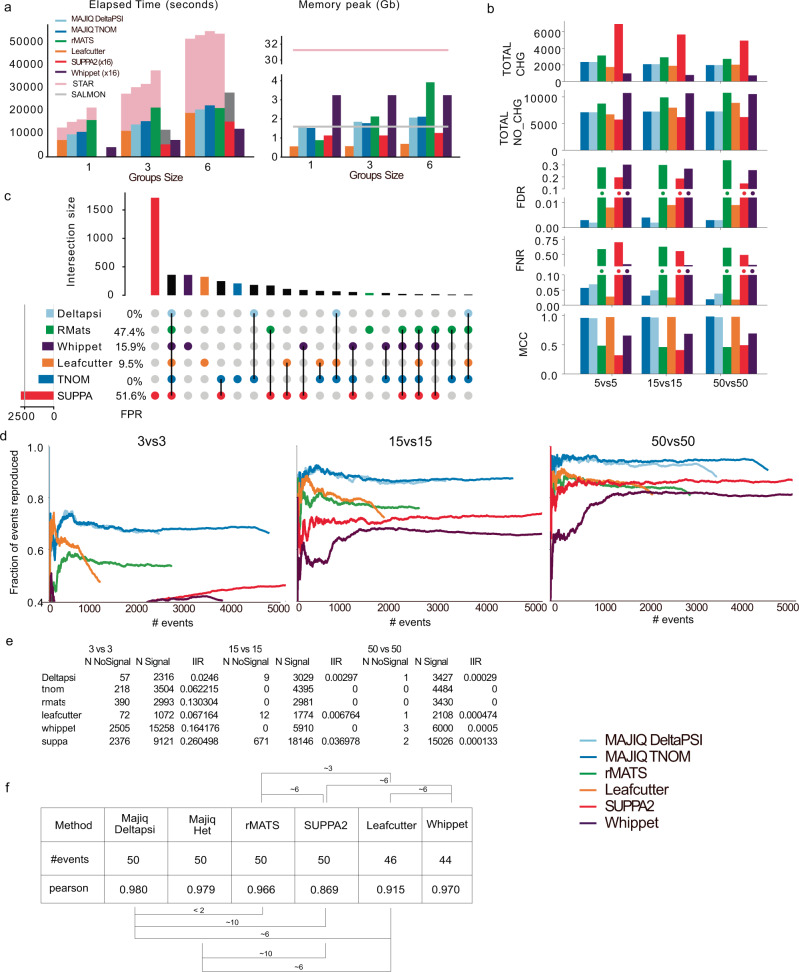
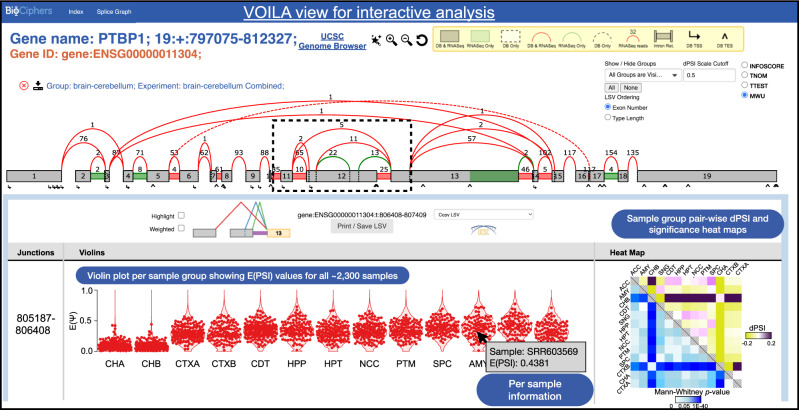
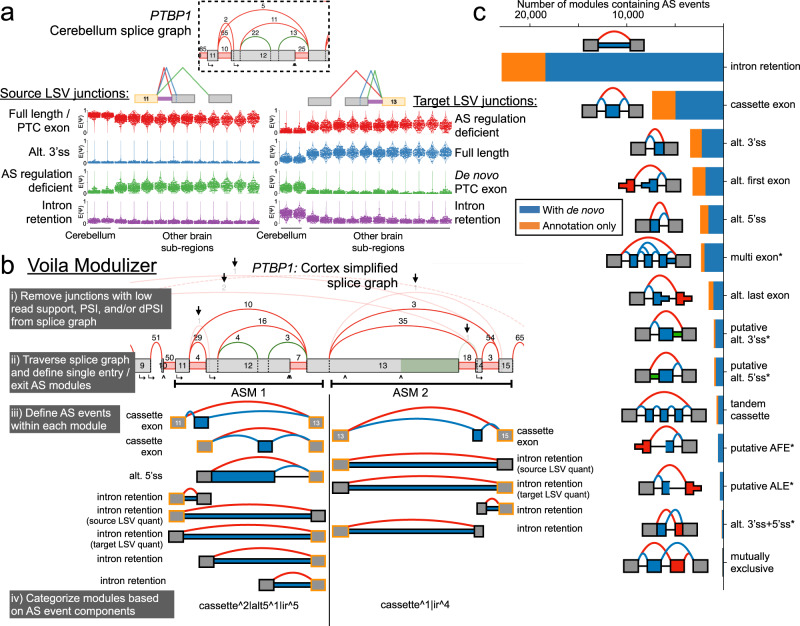
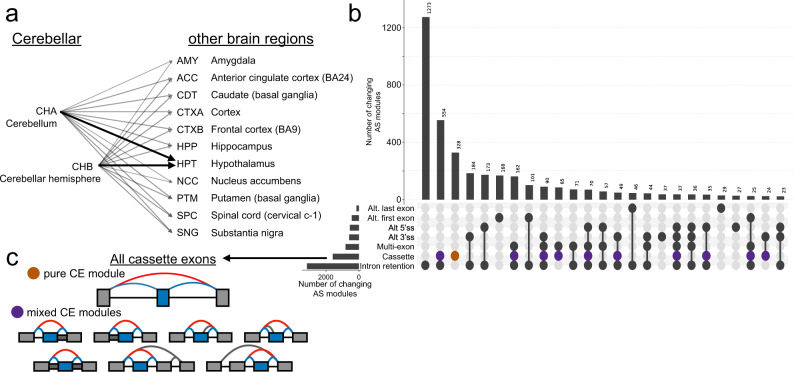
The ubiquity of RNA-seq has led to many methods that use RNA-seq data to analyze variations in RNA splicing. However, available methods are not well suited for handling heterogeneous and large datasets. Such datasets scale to thousands of samples across dozens of experimental conditions, exhibit increased variability compared to biological replicates, and involve thousands of unannotated splice variants resulting in increased transcriptome complexity. We describe here a suite of algorithms and tools implemented in the MAJIQ v2 package to address challenges in detection, quantification, and visualization of splicing variations from such datasets. Using both large scale synthetic data and GTEx v8 as benchmark datasets, we assess the advantages of MAJIQ v2 compared to existing methods. We then apply MAJIQ v2 package to analyze differential splicing across 2,335 samples from 13 brain subregions, demonstrating its ability to offer insights into brain subregion-specific splicing regulation.
RNA-seq 的广泛应用催生了许多利用 RNA-seq 数据来分析 RNA 剪接变异的方法。然而,现有的方法并不适合处理异构和大型数据集。这些数据集的规模达到了数千个样本,涉及数十个实验条件,与生物学重复相比,表现出更高的可变性,并且涉及数千个未注释的剪接变体,从而增加了转录组的复杂性。我们在这里描述了 MAJIQ v2 包中实现的一系列算法和工具,用于解决从这些数据集中检测、定量和可视化剪接变异的挑战。使用大规模的合成数据和 GTEx v8 作为基准数据集,我们评估了 MAJIQ v2 与现有方法相比的优势。然后,我们将 MAJIQ v2 包应用于分析来自 13 个大脑亚区的 2,335 个样本的差异剪接,展示了其提供大脑亚区特异性剪接调控见解的能力。