Faculty of Health Sciences, OsloMet - Oslo Metropolitan University, Oslo, Norway.
Department of Microbiology and Infection Control, Akershus University Hospital, Lørenskog, Norway.
Virol J. 2023 Mar 8;20(1):44. doi: 10.1186/s12985-023-02002-5.
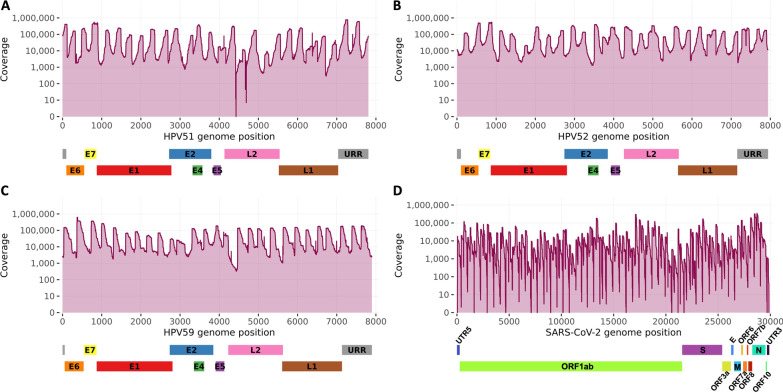
Previously developed TaME-seq method for deep sequencing of HPV, allowed simultaneous identification of the human papillomavirus (HPV) DNA consensus sequence, low-frequency variable sites, and chromosomal integration events. The method has been successfully validated and applied to the study of five carcinogenic high-risk (HR) HPV types (HPV16, 18, 31, 33, and 45). Here, we present TaME-seq2 with an updated laboratory workflow and bioinformatics pipeline. The HR-HPV type repertoire was expanded with HPV51, 52, and 59. As a proof-of-concept, TaME-seq2 was applied on SARS-CoV-2 positive samples showing the method's flexibility to a broader range of viruses, both DNA and RNA.
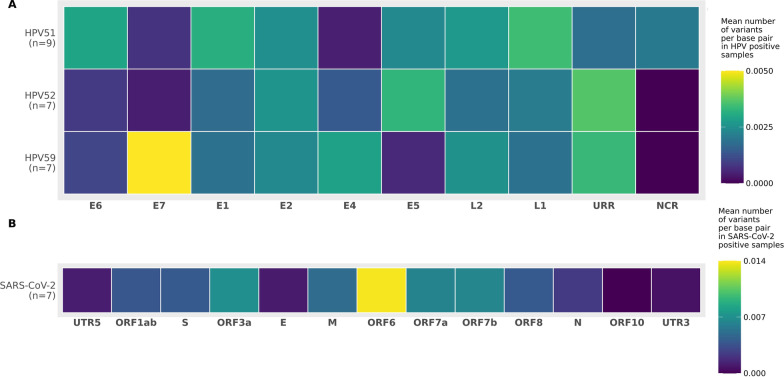
Compared to TaME-seq version 1, the bioinformatics pipeline of TaME-seq2 is approximately 40× faster. In total, 23 HPV-positive samples and seven SARS-CoV-2 clinical samples passed the threshold of 300× mean depth and were submitted to further analysis. The mean number of variable sites per 1 kb was ~ 1.5× higher in SARS-CoV-2 than in HPV-positive samples. Reproducibility and repeatability of the method were tested on a subset of samples. A viral integration breakpoint followed by a partial genomic deletion was found in within-run replicates of HPV59-positive sample. Identified viral consensus sequence in two separate runs was > 99.9% identical between replicates, differing by a couple of nucleotides identified in only one of the replicates. Conversely, the number of identical minor nucleotide variants (MNVs) differed greatly between replicates, probably caused by PCR-introduced bias. The total number of detected MNVs, calculated gene variability and mutational signature analysis, were unaffected by the sequencing run.
TaME-seq2 proved well suited for consensus sequence identification, and the detection of low-frequency viral genome variation and viral-chromosomal integrations. The repertoire of TaME-seq2 now encompasses seven HR-HPV types. Our goal is to further include all HR-HPV types in the TaME-seq2 repertoire. Moreover, with a minor modification of previously developed primers, the same method was successfully applied for the analysis of SARS-CoV-2 positive samples, implying the ease of adapting TaME-seq2 to other viruses.
先前开发的 TaME-seq 方法可用于 HPV 的深度测序,该方法允许同时鉴定人类乳头瘤病毒(HPV)的 DNA 共识序列、低频可变位点和染色体整合事件。该方法已成功验证并应用于五种致癌高危型(HR)HPV 类型(HPV16、18、31、33 和 45)的研究。在此,我们提出了 TaME-seq2,它具有更新的实验室工作流程和生物信息学管道。HR-HPV 型谱扩展到 HPV51、52 和 59。作为概念验证,TaME-seq2 应用于 SARS-CoV-2 阳性样本,表明该方法具有灵活性,可以应用于更广泛的 DNA 和 RNA 病毒。
与 TaME-seq 版本 1 相比,TaME-seq2 的生物信息学管道快约 40 倍。总共有 23 个 HPV 阳性样本和 7 个 SARS-CoV-2 临床样本通过了 300×平均深度的阈值,并进一步进行了分析。与 HPV 阳性样本相比,SARS-CoV-2 中每个 1kb 的可变位点数平均高约 1.5 倍。该方法的重复性和可重复性在样本的子集上进行了测试。在 HPV59 阳性样本的内部运行重复中发现了病毒整合断点和部分基因组缺失。在两个独立运行中鉴定的病毒共识序列在重复之间的相似度超过 99.9%,只有一个重复中鉴定出几个核苷酸的差异。相反,重复之间相同的次要核苷酸变异(MNV)数量差异很大,可能是由 PCR 引入的偏倚造成的。检测到的 MNV 总数、计算的基因变异性和突变特征分析不受测序运行的影响。
TaME-seq2 非常适合用于共识序列鉴定以及低频病毒基因组变异和病毒-染色体整合的检测。TaME-seq2 的型谱现在包括七种 HR-HPV 类型。我们的目标是进一步将所有 HR-HPV 类型纳入 TaME-seq2 型谱中。此外,通过对先前开发的引物进行微小修改,该方法成功应用于 SARS-CoV-2 阳性样本的分析,这意味着 TaME-seq2 很容易适应其他病毒。