Department of Global Health and Infectious Diseases, Brighton and Sussex Medical School, Brighton, United Kingdom.
Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
PLoS One. 2023 Mar 16;18(3):e0282584. doi: 10.1371/journal.pone.0282584. eCollection 2023.
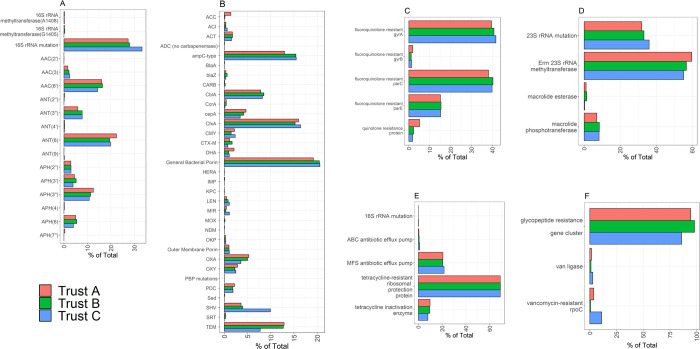
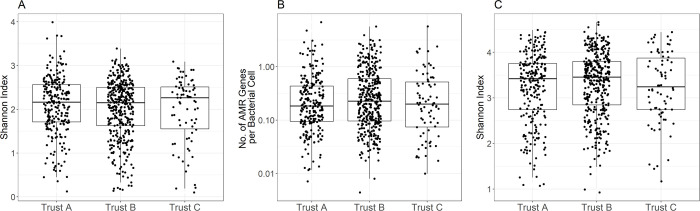
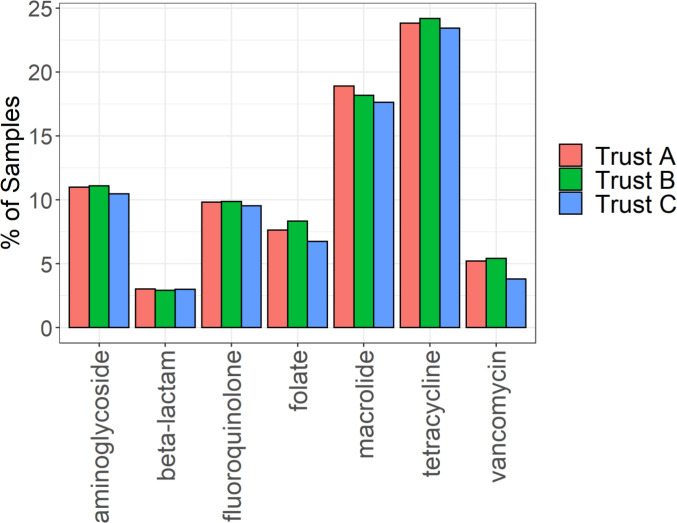
Antimicrobial resistance (AMR) is a threat to global public health. However, unsatisfactory approaches to directly measuring the AMR burden carried by individuals has hampered efforts to assess interventions aimed at reducing selection for AMR. Metagenomics can provide accurate detection and quantification of AMR genes within an individual person's faecal flora (their gut "resistome"). Using this approach, we aimed to test the hypothesis that differences in antimicrobial use across different hospitals in the United Kingdom will result in observable differences in the resistome of individual patients. Three National Health Service acute Hospital Trusts with markedly different antibiotic use and Clostridioides difficile infection rates collected faecal samples from anonymous patients which were discarded after C. difficile testing over a period of 9 to 15 months. Metagenomic DNA was extracted from these samples and sequenced using an Illumina NovaSeq 6000 platform. The resulting sequencing reads were analysed for taxonomic composition and for the presence of AMR genes. Among 683 faecal metagenomes we found huge variation between individuals in terms of taxonomic diversity (Shannon Index range 0.10-3.99) and carriage of AMR genes (Median 1.50 genes/cell/sample overall). We found no statistically significant differences in diversity (median Shannon index 2.16 (IQR 1.71-2.56), 2.15 (IQR 1.62-2.50) and 2.26 (IQR 1.55-2.51)) or carriage of AMR genes (median 1.37 genes/cell/sample (IQR 0.70-3.24), 1.70 (IQR 0.70-4.52) and 1.43 (IQR 0.55-3.71)) at the three trusts respectively. This was also the case across the sample collection period within the trusts. While we have not demonstrated differences over place or time using metagenomic sequencing of faecal discards, other sampling frameworks may be more suitable to determine whether organisational level differences in antibiotic use are associated with individual-level differences in burden of AMR carriage.
抗微生物药物耐药性(AMR)对全球公共卫生构成威胁。然而,由于人们不满意直接衡量个体携带的 AMR 负担的方法,因此评估旨在减少 AMR 选择的干预措施的工作受到了阻碍。宏基因组学可以在个体的粪便菌群(他们的肠道“耐药组”)中准确检测和定量 AMR 基因。使用这种方法,我们旨在检验以下假设:英国不同医院之间使用的抗菌药物存在差异,这将导致个体患者的耐药组出现可观察到的差异。三家具有明显不同抗生素使用和艰难梭菌感染率的英国国民保健制度急性医院信托机构从匿名患者那里收集粪便样本,在艰难梭菌检测后的 9 至 15 个月期间,这些样本被丢弃。从这些样本中提取宏基因组 DNA 并使用 Illumina NovaSeq 6000 平台进行测序。对所得测序读长进行分析,以分析分类组成和 AMR 基因的存在情况。在 683 个粪便宏基因组中,我们发现个体之间在分类多样性(香农指数范围 0.10-3.99)和 AMR 基因携带方面存在巨大差异(总体中位数为 1.50 个基因/细胞/样本)。我们没有发现多样性(中位数香农指数 2.16(IQR 1.71-2.56)、2.15(IQR 1.62-2.50)和 2.26(IQR 1.55-2.51))或 AMR 基因携带(中位数 1.37 个基因/细胞/样本(IQR 0.70-3.24)、1.70(IQR 0.70-4.52)和 1.43(IQR 0.55-3.71))方面存在统计学上无显著差异在三个信托基金中分别。在信托机构内部的样本采集期间,情况也是如此。虽然我们没有通过对粪便丢弃物的宏基因组测序来证明位置或时间上的差异,但其他采样框架可能更适合确定抗生素使用的组织水平差异是否与 AMR 携带负担的个体水平差异相关。