Sullivan Patrick F, Meadows Jennifer R S, Gazal Steven, Phan BaDoi N, Li Xue, Genereux Diane P, Dong Michael X, Bianchi Matteo, Andrews Gregory, Sakthikumar Sharadha, Nordin Jessika, Roy Ananya, Christmas Matthew J, Marinescu Voichita D, Wallerman Ola, Xue James R, Li Yun, Yao Shuyang, Sun Quan, Szatkiewicz Jin, Wen Jia, Huckins Laura M, Lawler Alyssa J, Keough Kathleen C, Zheng Zhili, Zeng Jian, Wray Naomi R, Johnson Jessica, Chen Jiawen, Paten Benedict, Reilly Steven K, Hughes Graham M, Weng Zhiping, Pollard Katherine S, Pfenning Andreas R, Forsberg-Nilsson Karin, Karlsson Elinor K, Lindblad-Toh Kerstin
Department of Genetics, University of North Carolina Medical School; Chapel Hill, NC 27599, USA.
Department of Medical Epidemiology and Biostatistics, Karolinska Institutet; Stockholm, Sweden.
bioRxiv. 2023 Mar 10:2023.03.10.531987. doi: 10.1101/2023.03.10.531987.
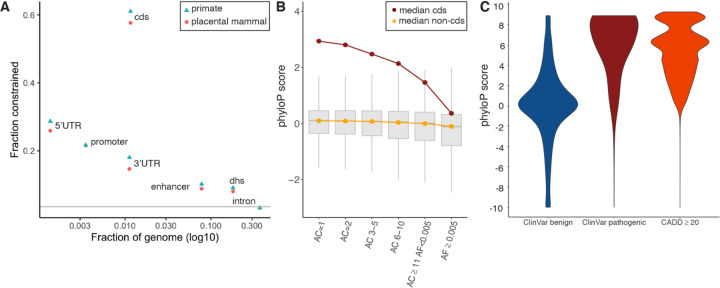
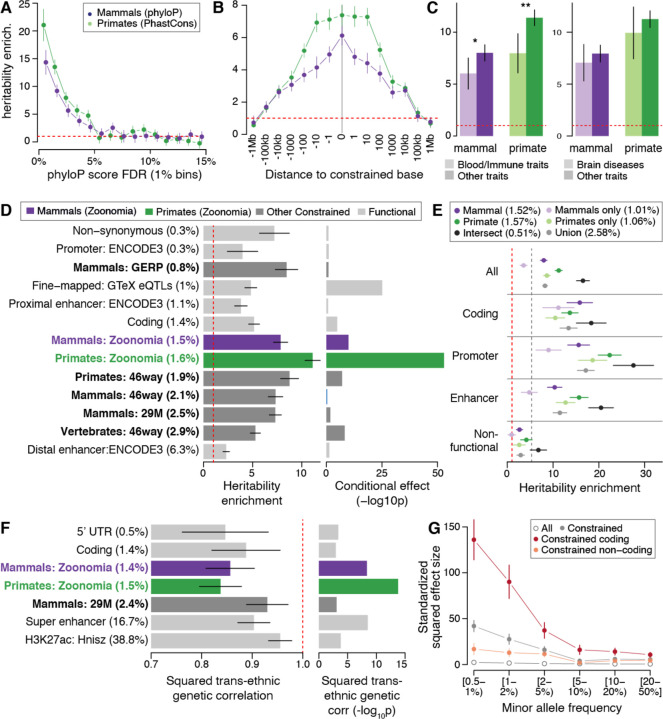
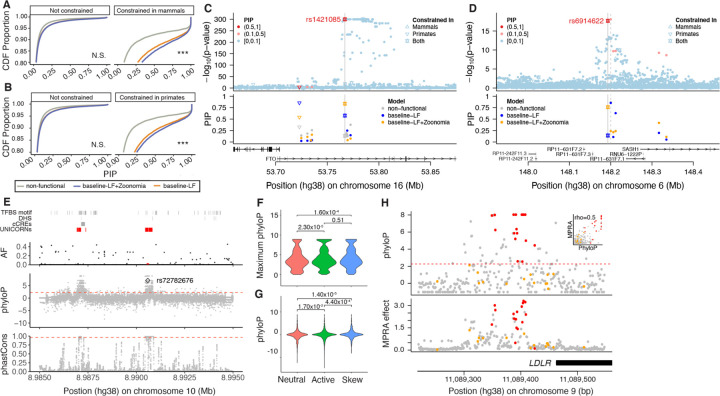
Although thousands of genomic regions have been associated with heritable human diseases, attempts to elucidate biological mechanisms are impeded by a general inability to discern which genomic positions are functionally important. Evolutionary constraint is a powerful predictor of function that is agnostic to cell type or disease mechanism. Here, single base phyloP scores from the whole genome alignment of 240 placental mammals identified 3.5% of the human genome as significantly constrained, and likely functional. We compared these scores to large-scale genome annotation, genome-wide association studies (GWAS), copy number variation, clinical genetics findings, and cancer data sets. Evolutionarily constrained positions are enriched for variants explaining common disease heritability (more than any other functional annotation). Our results improve variant annotation but also highlight that the regulatory landscape of the human genome still needs to be further explored and linked to disease.
尽管数千个基因组区域已与人类遗传性疾病相关联,但由于普遍无法辨别哪些基因组位置具有功能重要性,阐明生物学机制的尝试受到了阻碍。进化约束是功能的有力预测指标,与细胞类型或疾病机制无关。在这里,来自240种胎盘哺乳动物全基因组比对的单碱基phyloP评分确定了人类基因组的3.5%受到显著约束且可能具有功能。我们将这些评分与大规模基因组注释、全基因组关联研究(GWAS)、拷贝数变异、临床遗传学发现和癌症数据集进行了比较。进化上受约束的位置富含解释常见疾病遗传力的变异(比任何其他功能注释都多)。我们的结果改进了变异注释,但也突出表明人类基因组的调控格局仍需进一步探索并与疾病建立联系。