Institut für Informationsverarbeitung and L3S Research Center, Leibniz University Hannover, Hannover, Germany.
Institut für Nachrichtentechnik, RWTH Aachen University, Aachen, Germany.
BMC Bioinformatics. 2023 Mar 28;24(1):121. doi: 10.1186/s12859-023-05240-0.
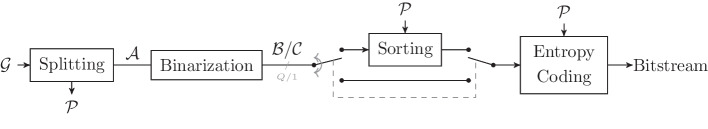
In recent years, advances in high-throughput sequencing technologies have enabled the use of genomic information in many fields, such as precision medicine, oncology, and food quality control. The amount of genomic data being generated is growing rapidly and is expected to soon surpass the amount of video data. The majority of sequencing experiments, such as genome-wide association studies, have the goal of identifying variations in the gene sequence to better understand phenotypic variations. We present a novel approach for compressing gene sequence variations with random access capability: the Genomic Variant Codec (GVC). We use techniques such as binarization, joint row- and column-wise sorting of blocks of variations, as well as the image compression standard JBIG for efficient entropy coding.
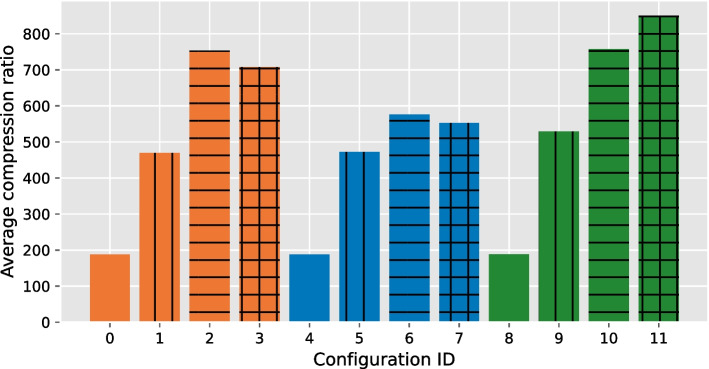
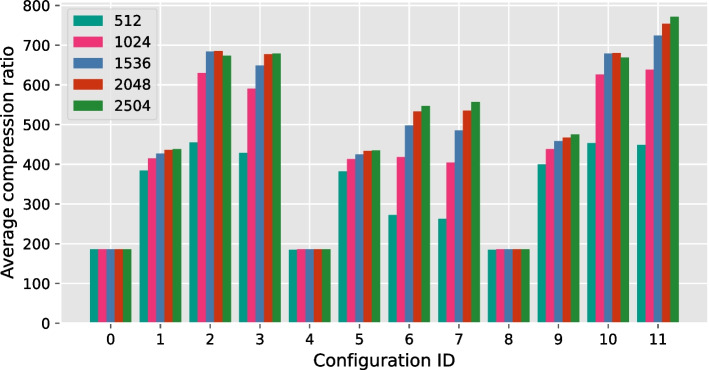
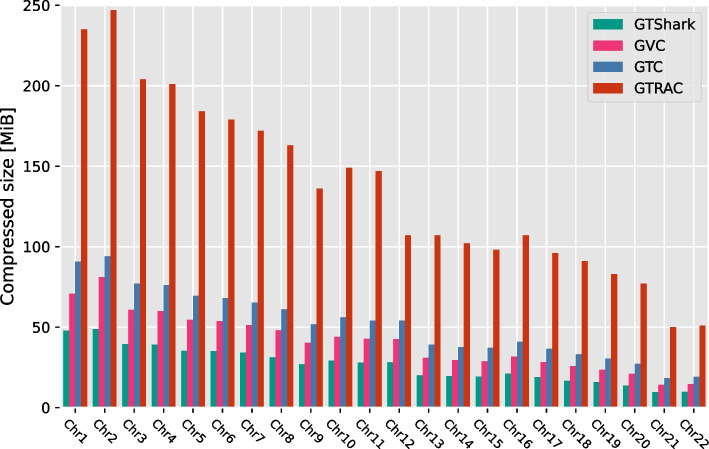
Our results show that GVC provides the best trade-off between compression and random access compared to the state of the art: it reduces the genotype information size from 758 GiB down to 890 MiB on the publicly available 1000 Genomes Project (phase 3) data, which is 21% less than the state of the art in random-access capable methods.
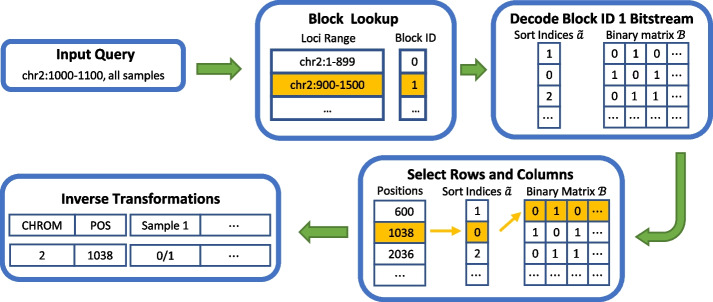
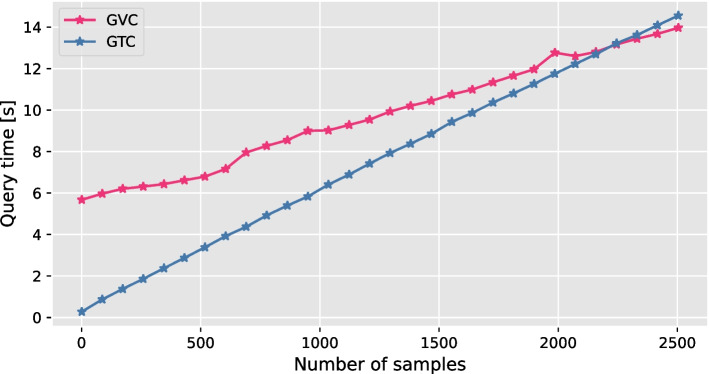
By providing the best results in terms of combined random access and compression, GVC facilitates the efficient storage of large collections of gene sequence variations. In particular, the random access capability of GVC enables seamless remote data access and application integration. The software is open source and available at https://github.com/sXperfect/gvc/ .
近年来,高通量测序技术的进步使得基因组信息在许多领域得到了应用,如精准医学、肿瘤学和食品质量控制。生成的基因组数据量正在迅速增长,预计很快将超过视频数据量。大多数测序实验,如全基因组关联研究,旨在识别基因序列中的变异,以更好地理解表型变异。我们提出了一种具有随机访问能力的基因序列变异压缩的新方法:基因组变异编解码器(GVC)。我们使用了二进制化、块的行和列联合排序以及 JBIG 图像压缩标准等技术,以实现高效的熵编码。
我们的结果表明,与最先进的方法相比,GVC 在压缩和随机访问之间提供了最佳的折衷:它将公开可用的 1000 基因组计划(第 3 阶段)数据中的基因型信息大小从 758 GiB 减少到 890 MiB,比具有随机访问能力的方法减少了 21%。
通过在随机访问和压缩方面提供最佳的结果,GVC 促进了大规模基因序列变异的高效存储。特别是,GVC 的随机访问能力实现了无缝的远程数据访问和应用程序集成。该软件是开源的,可在 https://github.com/sXperfect/gvc/ 获得。