Division of Systems Medicine, School of Medicine, Ninewells Hospital and Medical School, Dundee, UK.
Wellcome-MRC Institute of Metabolic Science, University of Cambridge, Cambridge, UK.
Diabetologia. 2023 Jul;66(7):1340-1352. doi: 10.1007/s00125-023-05907-6. Epub 2023 Apr 4.
AIMS/HYPOTHESIS: Chronic hyperglycaemia and recurrent hypoglycaemia are independently associated with accelerated cognitive decline in type 1 diabetes. Recurrent hypoglycaemia in rodent models of chemically induced (streptozotocin [STZ]) diabetes leads to cognitive impairment in memory-related tasks associated with hippocampal oxidative damage. This study examined the hypothesis that post-hypoglycaemic hyperglycaemia in STZ-diabetes exacerbates hippocampal oxidative stress and explored potential contributory mechanisms.
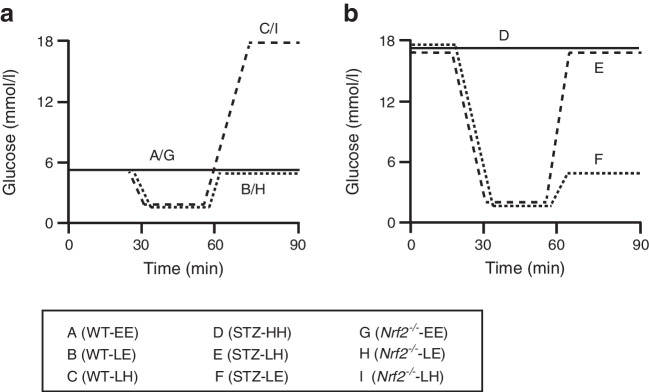
The hyperinsulinaemic glucose clamp technique was used to induce equivalent hypoglycaemia and to control post-hypoglycaemic glucose levels in mice with and without STZ-diabetes and Nrf2 mice (lacking Nrf2 [also known as Nfe2l2]). Subsequently, quantitative proteomics based on stable isotope labelling by amino acids in cell culture and biochemical approaches were used to assess oxidative damage and explore contributory pathways.
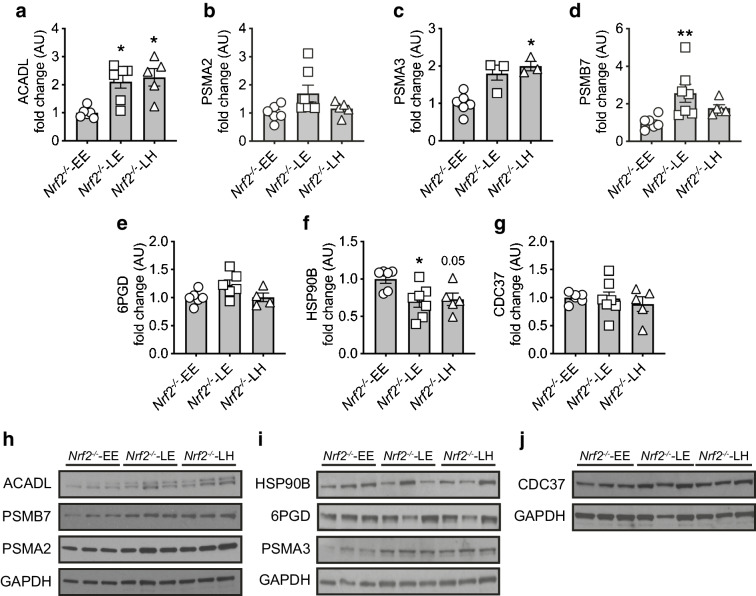
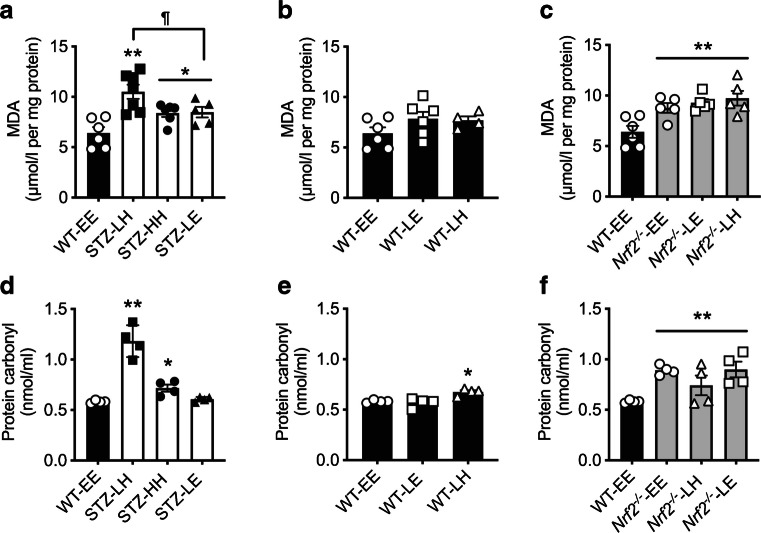
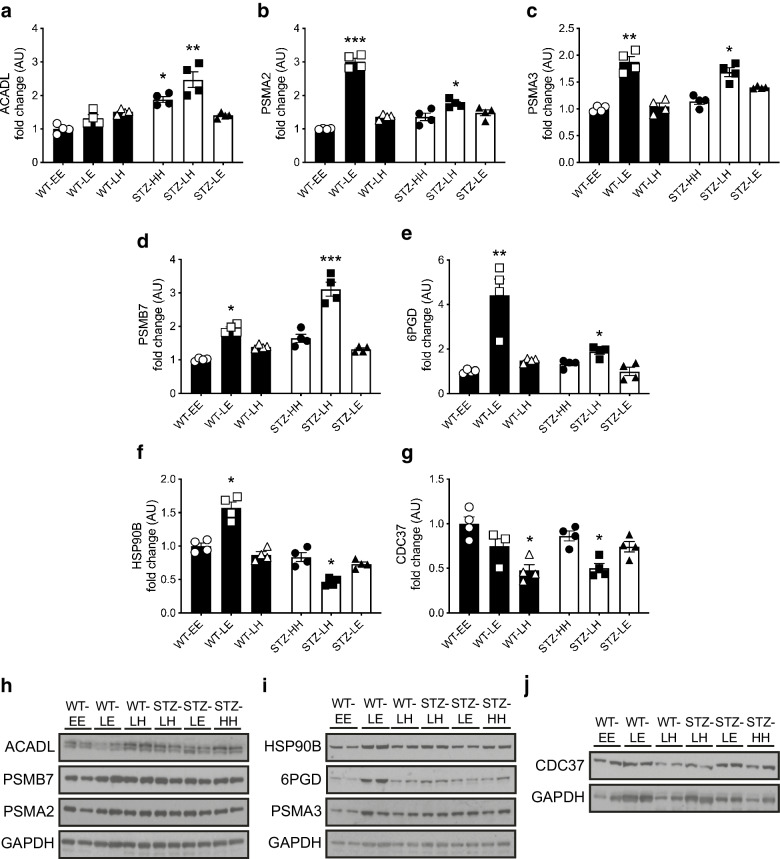
Evidence of hippocampal oxidative damage was most marked in mice with STZ-diabetes exposed to post-hypoglycaemic hyperglycaemia; these mice also showed induction of Nrf2 and the Nrf2 transcriptional targets Sod2 and Hmox-1. In this group, hypoglycaemia induced a significant upregulation of proteins involved in alternative fuel provision, reductive biosynthesis and degradation of damaged proteins, and a significant downregulation of proteins mediating the stress response. Key differences emerged between mice with and without STZ-diabetes following recovery from hypoglycaemia in proteins mediating the stress response and reductive biosynthesis.
CONCLUSIONS/INTERPRETATION: There is a disruption of the cellular response to a hypoglycaemic challenge in mice with STZ-induced diabetes that is not seen in wild-type non-diabetic animals. The chronic hyperglycaemia of diabetes and post-hypoglycaemic hyperglycaemia act synergistically to induce oxidative stress and damage in the hippocampus, possibly leading to irreversible damage/modification to proteins or synapses between cells. In conclusion, recurrent hypoglycaemia in sub-optimally controlled diabetes may contribute, at least in part, to accelerated cognitive decline through amplifying oxidative damage in key brain regions, such as the hippocampus.
The datasets generated during and/or analysed during the current study are available in ProteomeXchange, accession no. 1-20220824-173727 ( www.proteomexchange.org ). Additional datasets generated during and/or analysed during the present study are available from the corresponding author upon reasonable request.
目的/假设:慢性高血糖和反复低血糖与 1 型糖尿病患者认知能力加速下降独立相关。化学诱导(链脲佐菌素[STZ])糖尿病的啮齿动物模型中的反复低血糖会导致与海马氧化损伤相关的记忆相关任务的认知障碍。本研究检验了以下假设:STZ 糖尿病患者的低血糖后高血糖会加剧海马氧化应激,并探讨了潜在的促成机制。
使用高胰岛素葡萄糖钳夹技术在有和没有 STZ 糖尿病和 Nrf2 小鼠(缺乏 Nrf2[也称为 Nfe2l2])的情况下诱导等效低血糖,并控制低血糖后的血糖水平。随后,基于细胞培养中稳定同位素标记的氨基酸的定量蛋白质组学和生化方法用于评估氧化损伤并探索促成途径。
在暴露于低血糖后高血糖的 STZ 糖尿病小鼠中,海马氧化损伤的证据最为明显;这些小鼠还表现出 Nrf2 和 Nrf2 转录靶标 Sod2 和 Hmox-1 的诱导。在该组中,低血糖诱导了参与替代燃料供应、还原性生物合成和受损蛋白质降解的蛋白质的显著上调,以及介导应激反应的蛋白质的显著下调。在从低血糖中恢复后,STZ 糖尿病小鼠与非糖尿病野生型小鼠之间在介导应激反应和还原性生物合成的蛋白质方面出现了关键差异。
结论/解释:在 STZ 诱导的糖尿病小鼠中,对低血糖挑战的细胞反应出现了中断,而在非糖尿病野生型动物中则没有观察到这种中断。糖尿病的慢性高血糖和低血糖后高血糖协同作用,导致海马氧化应激和损伤,可能导致细胞间蛋白质或突触不可逆的损伤/修饰。总之,在控制不佳的糖尿病中反复发生的低血糖可能会通过放大关键大脑区域(如海马体)中的氧化损伤,至少部分导致认知能力加速下降。
本研究期间产生和/或分析的数据可在 ProteomeXchange 中获得,注册号为 1-20220824-173727(www.proteomexchange.org)。本研究期间产生和/或分析的其他数据集可应合理要求向通讯作者索取。