Liferiver Science and Technology Institute, Shanghai ZJ Bio-Tech Co., Ltd., Shanghai, China.
BMC Bioinformatics. 2023 Apr 4;24(1):128. doi: 10.1186/s12859-023-05195-2.
Concentrations of the pathogenic microorganisms' DNA in biological samples are typically low. Therefore, DNA diagnostics of common infections are costly, rarely accurate, and challenging. Limited by failing to cover updated epidemic testing samples, computational services are difficult to implement in clinical applications without complex customized settings. Furthermore, the combined biomarkers used to maintain high conservation may not be cost effective and could cause several experimental errors in many clinical settings. Given the limitations of recent developed technology, 16S rRNA is too conserved to distinguish closely related species, and mosaic plasmids are not effective as well because of their uneven distribution across prokaryotic taxa.
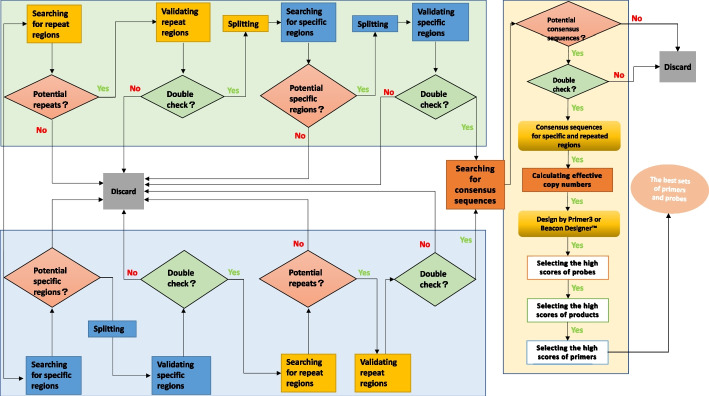
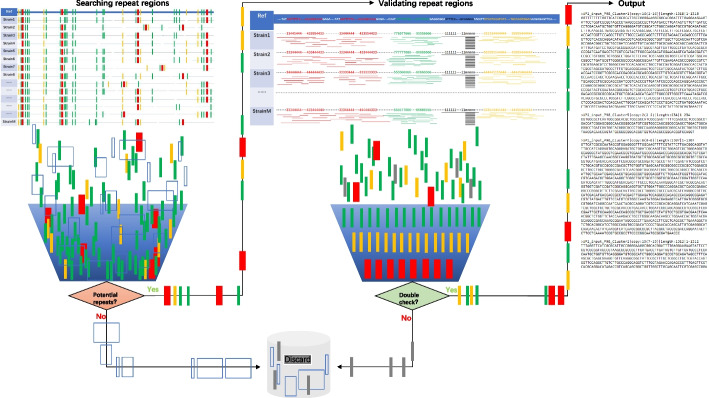
Here, we provide a computational strategy, Shine, that allows extraction of specific, sensitive and well-conserved biomarkers from massive microbial genomic datasets. Distinguished with simple concatenations with blast-based filtering, our method involves a de novo genome alignment-based pipeline to explore the original and specific repetitive biomarkers in the defined population. It can cover all members to detect newly discovered multicopy conserved species-specific or even subspecies-specific target probes and primer sets. The method has been successfully applied to a number of clinical projects and has the overwhelming advantages of automated detection of all pathogenic microorganisms without the limitations of genome annotation and incompletely assembled motifs. Using on our pipeline, users may select different configuration parameters depending on the purpose of the project for routine clinical detection practices on the website https://bioinfo.liferiver.com.cn with easy registration.
The proposed strategy is suitable for identifying shared phylogenetic markers while featuring low rates of false positive or false negative. This technology is suitable for the automatic design of minimal and efficient PCR primers and other types of detection probes.
生物样本中致病微生物 DNA 的浓度通常较低。因此,常见感染的 DNA 诊断既昂贵,又不准确,还具有挑战性。由于计算服务无法涵盖最新的流行测试样本,因此在没有复杂定制设置的情况下,很难在临床应用中实施。此外,用于保持高保守性的组合生物标志物可能不具有成本效益,并且在许多临床环境中可能会导致多个实验错误。鉴于最近开发的技术存在局限性,16S rRNA 由于过于保守,无法区分密切相关的物种,而嵌合质粒也因为在原核生物分类中分布不均匀而效果不佳。
在这里,我们提供了一种计算策略 Shine,可以从大量微生物基因组数据集中提取特定、敏感且高度保守的生物标志物。与基于 Blast 的过滤的简单串联区分开来,我们的方法涉及一种基于从头基因组比对的管道,用于探索定义种群中的原始和特定重复生物标志物。它可以涵盖所有成员,以检测新发现的多拷贝保守物种特异性甚至亚种特异性目标探针和引物对。该方法已成功应用于多个临床项目,具有自动化检测所有致病微生物的压倒性优势,而不受基因组注释和不完全组装基序的限制。用户可以根据项目的目的,在网站 https://bioinfo.liferiver.com.cn 上使用我们的管道选择不同的配置参数,方便注册,进行常规临床检测。
所提出的策略适合识别共享的系统发育标记,同时具有较低的假阳性或假阴性率。该技术适用于最小化和有效 PCR 引物等其他类型检测探针的自动设计。