Mori Mari, Clause Amanda R, Truxal Kristen, Hagelstrom R Tanner, Manickam Kandamurugu, Kaler Stephen G, Prasad Vinay, Windster Jonathan, Alves Maria M, Di Lorenzo Carlo
From the Division of Genetic and Genomic Medicine, Nationwide Children's Hospital, Columbus, OH.
Department of Pediatrics, The Ohio State University, Columbus, OH.
JPGN Rep. 2022 Oct 20;3(4):e258. doi: 10.1097/PG9.0000000000000258. eCollection 2022 Nov.
Pediatric intestinal pseudo-obstruction (PIPO) is a heterogeneous condition characterized by impaired gastrointestinal propulsion, a broad clinical spectrum, and variable severity. Several molecular bases underlying primary PIPO have been identified, of which autosomal dominant ACTG2-related visceral myopathy is the most common in both familial or sporadic primary PIPO cases. We present a family with autosomal recessive ACTG2-related disease in which both parents have mild gastrointestinal symptoms and sons have severe PIPO and bladder dysfunction.
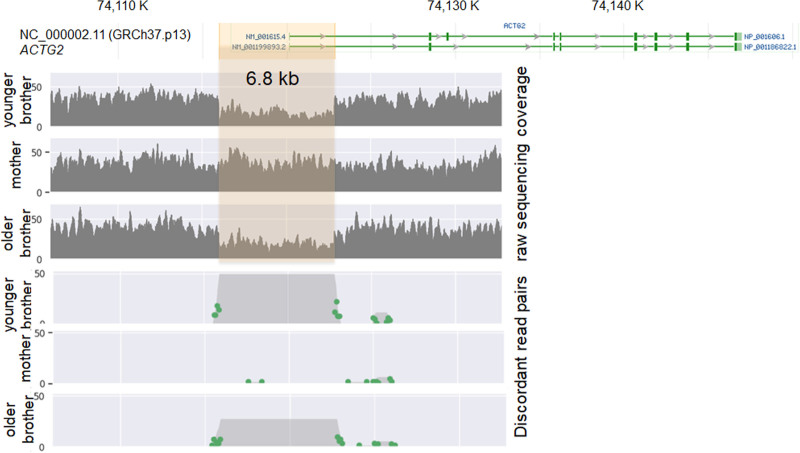
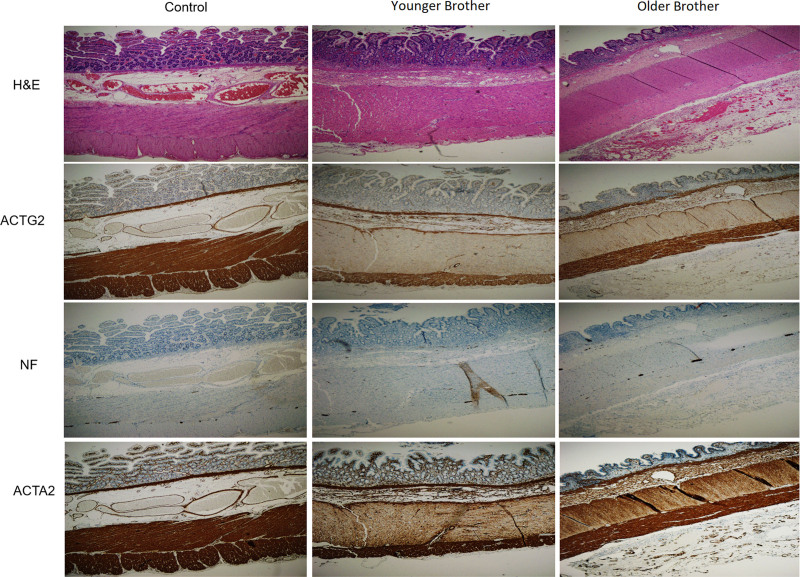
Clinical genome sequencing was performed on the patients and the mother. Immunohistochemistry was performed on intestinal tissue from the patients to show expression levels of the .
Genome sequencing identified a 6.8 kb 2p13.1 loss that includes the gene and a maternally inherited missense variant p.Val10Met in the gene.
This case demonstrates that monoallelic hypomorphic variants may underly mild primary gastrointestinal symptoms, while biallelic mild variants can cause severe diseases. The Deletions of the noncoding exon can be an under-recognized cause of mild gastrointestinal symptoms unidentifiable by exome sequencing, explaining some instances of interfamilial variability with an apparent autosomal dominant inheritance. Genome sequencing is recommended as a genetic work-up for primary or idiopathic PIPO because of genetic heterogeneity.
小儿肠假性梗阻(PIPO)是一种异质性疾病,其特征为胃肠推进功能受损、临床谱广且严重程度各异。已确定了原发性PIPO的几种分子基础,其中常染色体显性ACTG2相关内脏肌病在家族性或散发性原发性PIPO病例中最为常见。我们报告了一个患有常染色体隐性ACTG2相关疾病的家族,其中父母均有轻度胃肠道症状,儿子患有严重的PIPO和膀胱功能障碍。
对患者及其母亲进行临床基因组测序。对患者的肠道组织进行免疫组织化学检测以显示……的表达水平。
基因组测序确定了一个6.8kb的2p13.1缺失,其中包括……基因以及该基因中一个母系遗传的错义变异p.Val10Met。
该病例表明,单等位基因低表达变异可能是轻度原发性胃肠道症状的基础,而双等位基因轻度变异可导致严重疾病。非编码外显子的缺失可能是外显子测序无法识别的轻度胃肠道症状的一个未被充分认识的原因,这解释了一些具有明显常染色体显性遗传的家族间变异性情况。由于遗传异质性,建议将基因组测序作为原发性或特发性PIPO的基因检查方法。