Haematology Unit, Cancer Research Centre, Institute for Medical Research, National Institutes of Health, Ministry of Health Malaysia, 40170, Shah Alam, Selangor, Malaysia.
Centre for Medical Laboratory Technology Studies, Faculty of Health Sciences, Universiti Teknologi MARA, 42300, Puncak Alam, Selangor, Malaysia.
J Med Case Rep. 2023 Jun 10;17(1):250. doi: 10.1186/s13256-023-03984-0.
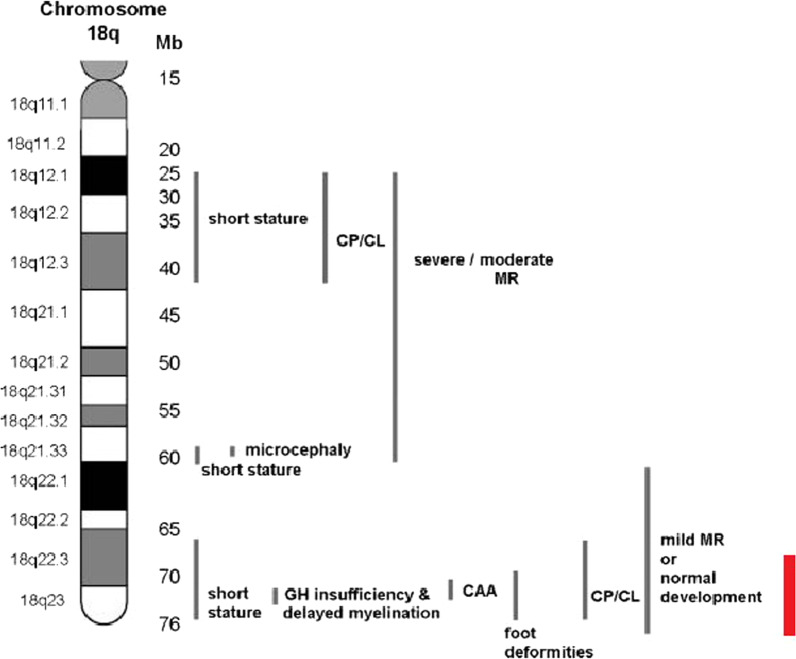
The 18q- deletion syndrome is a rare congenital chromosomal disorder caused by a partial deletion of the long arm of chromosome 18. The diagnosis of a patient with this syndrome relies on the family medical history, physical examination, developmental assessment, and cytogenetic findings. However, the phenotype of patients with 18q- deletion syndrome can be highly variable, ranging from almost normal to severe malformations and intellectual disability, and normal cytogenetic findings are common, thus complicating the diagnosis. Interestingly, only few characteristic features of typical 18q- deletion syndrome were found in the patient, despite sharing the same critical region. To our knowledge, this is the first report of a Malaysian individual with 18q- terminal microdeletion diagnosed with microarray-based technology.
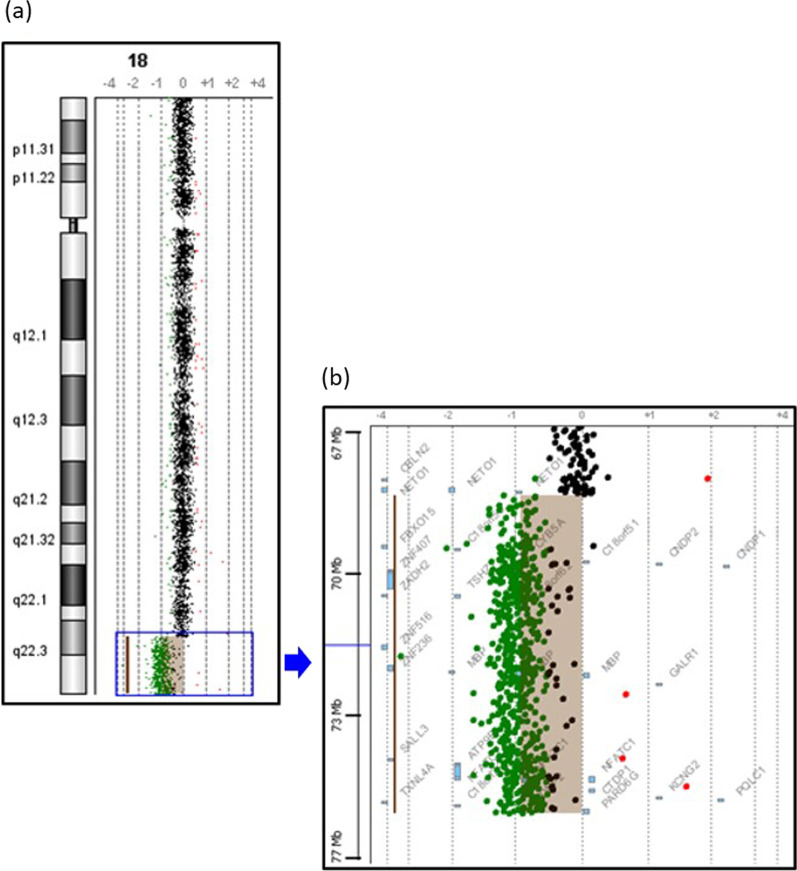
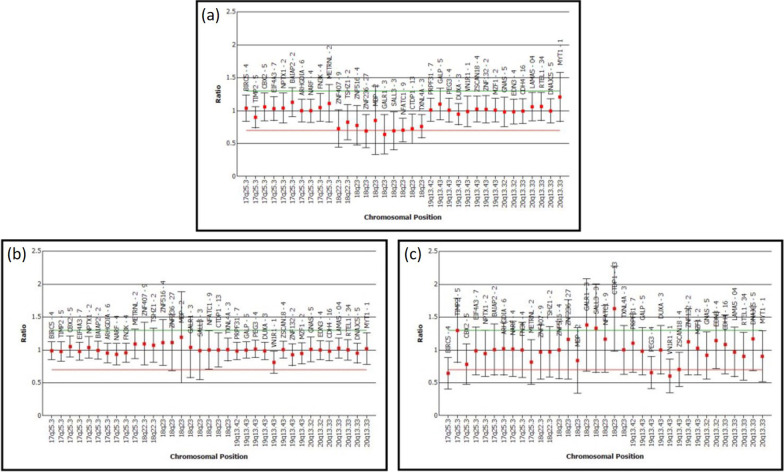
Here we report a 16-year-old Malaysian Chinese boy, a product of a non-consanguineous marriage, who presented with intellectual disability, facial dysmorphism, high arched palate, congenital talipes equinovarus (clubfoot), congenital scoliosis, congenital heart defect, and behavioral problems. A routine chromosome analysis on 20 metaphase cells showed a normal 46, XY G-banded karyotype. Array-based comparative genomic hybridization was performed using a commercially available 244 K 60-mer oligonucleotide microarray slide according to the manufacturer's protocol. This platform allows genome-wide survey and molecular profiling of genomic aberrations with an average resolution of about 10 kB. In addition, multiplex ligation-dependent probe amplification analysis was carried out using SALSA MLPA kit P320 Telomere-13 to confirm the array-based comparative genomic hybridization finding. Array-based comparative genomic hybridization analysis revealed a 7.3 MB terminal deletion involving chromosome band 18q22.3-qter. This finding was confirmed by multiplex ligation-dependent probe amplification, where a deletion of ten probes mapping to the 18q22.3-q23 region was detected, and further multiplex ligation-dependent probe amplification analysis on his parents showed the deletion to be de novo.
The findings from this study expand the phenotypic spectrum of the 18q- deletion syndrome by presenting a variation of typical 18q- deletion syndrome features to the literature. In addition, this case report demonstrated the ability of the molecular karyotyping method, such as array-based comparative genomic hybridization, to assist in the diagnosis of cases with a highly variable phenotype and variable aberrations, such as 18q- deletion syndrome.
18q- 缺失综合征是一种罕见的先天性染色体疾病,由染色体 18 长臂的部分缺失引起。该综合征患者的诊断依赖于家族病史、体格检查、发育评估和细胞遗传学发现。然而,18q- 缺失综合征患者的表型可能高度可变,从几乎正常到严重畸形和智力障碍不等,且常见正常细胞遗传学发现,从而使诊断变得复杂。有趣的是,尽管共享相同的关键区域,但该患者仅发现了典型 18q- 缺失综合征的少数特征。据我们所知,这是首例使用基于微阵列的技术诊断马来西亚个体 18q- 末端微缺失的报告。
我们在此报告了一名 16 岁的马来西亚华裔男孩,他是非近亲结婚的产物,患有智力障碍、面部畸形、高拱形腭、先天性马蹄内翻足(马蹄足)、先天性脊柱侧凸、先天性心脏病和行为问题。对 20 个中期细胞进行常规染色体分析显示正常的 46,XY G 带核型。根据制造商的方案,使用商业上可用的 244K 60-mer 寡核苷酸微阵列玻片进行基于微阵列的比较基因组杂交。该平台允许对基因组畸变进行全基因组检测和分子分析,平均分辨率约为 10KB。此外,使用 SALSA MLPA 试剂盒 P320 Telomere-13 进行多重连接依赖性探针扩增分析,以确认基于微阵列的比较基因组杂交结果。基于微阵列的比较基因组杂交分析显示,染色体 18q22.3-qter 存在 7.3MB 的末端缺失。通过多重连接依赖性探针扩增检测到十个映射到 18q22.3-q23 区域的探针缺失,进一步对其父母进行多重连接依赖性探针扩增分析显示该缺失为新生。
本研究通过向文献呈现典型 18q- 缺失综合征特征的变化,扩展了 18q- 缺失综合征的表型谱。此外,该病例报告证明了分子核型分析方法(如基于微阵列的比较基因组杂交)的能力,可协助诊断表型高度可变和异常多变的病例,如 18q- 缺失综合征。