MyOme Inc., 535 Middlefield Rd Suite 170, Menlo Park, CA, USA.
Department of Pathology and Cell Biology, Columbia University Irving Medical Center, New York, NY, USA.
J Transl Med. 2023 Jun 10;21(1):378. doi: 10.1186/s12967-023-04243-y.
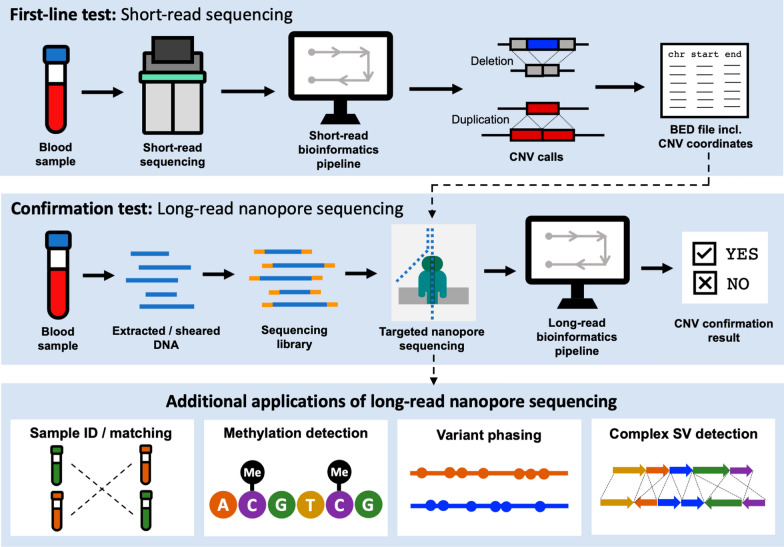
Diagnosis of rare genetic diseases can be a long, expensive and complex process, involving an array of tests in the hope of obtaining an actionable result. Long-read sequencing platforms offer the opportunity to make definitive molecular diagnoses using a single assay capable of detecting variants, characterizing methylation patterns, resolving complex rearrangements, and assigning findings to long-range haplotypes. Here, we demonstrate the clinical utility of Nanopore long-read sequencing by validating a confirmatory test for copy number variants (CNVs) in neurodevelopmental disorders and illustrate the broader applications of this platform to assess genomic features with significant clinical implications.
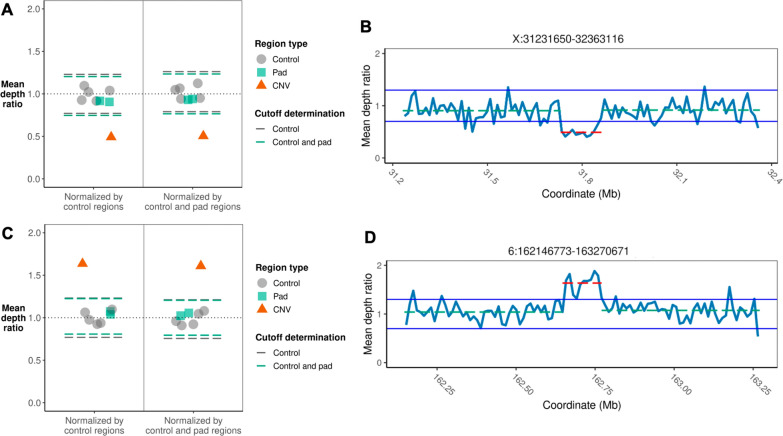
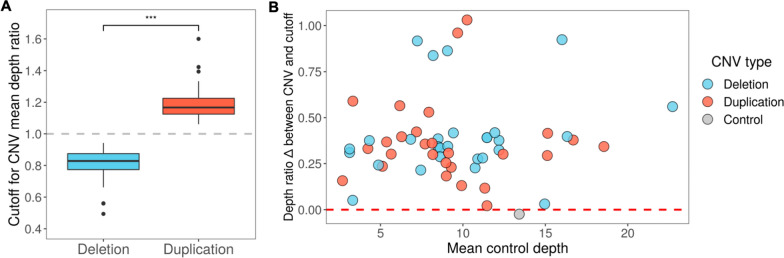
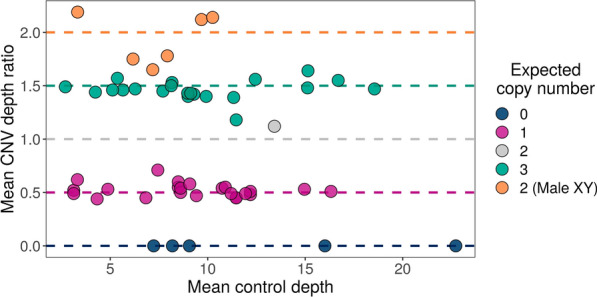
We used adaptive sampling on the Oxford Nanopore platform to sequence 25 genomic DNA samples and 5 blood samples collected from patients with known or false-positive copy number changes originally detected using short-read sequencing. Across the 30 samples (a total of 50 with replicates), we assayed 35 known unique CNVs (a total of 55 with replicates) and one false-positive CNV, ranging in size from 40 kb to 155 Mb, and assessed the presence or absence of suspected CNVs using normalized read depth.
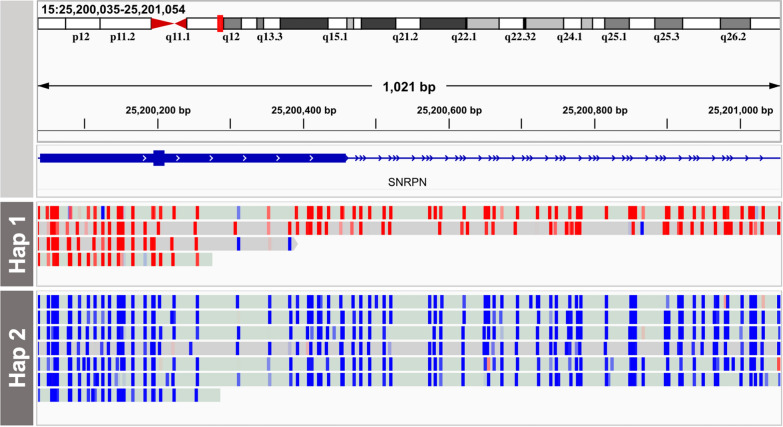
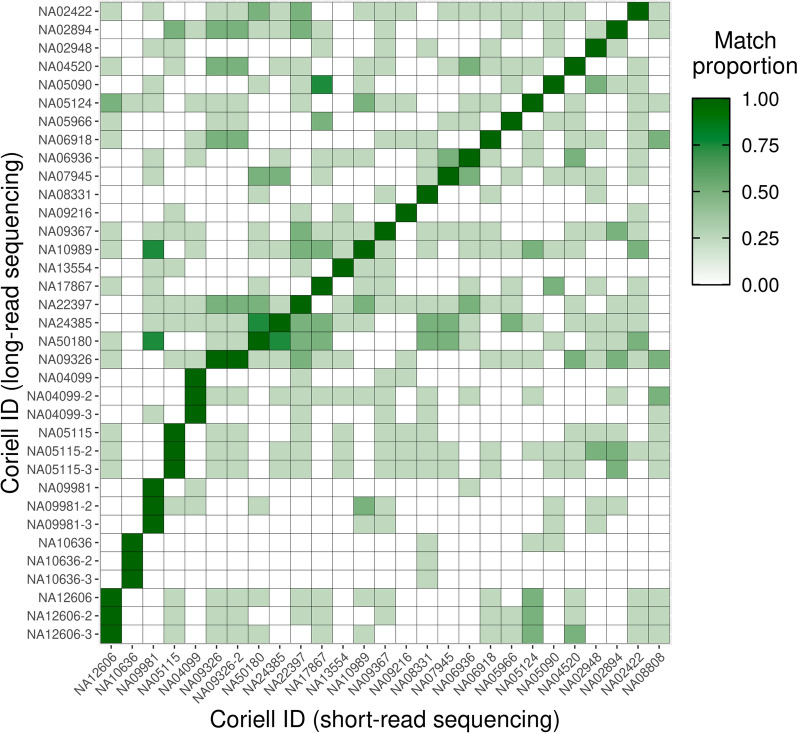
Across 50 samples (including replicates) sequenced on individual MinION flow cells, we achieved an average on-target mean depth of 9.5X and an average on-target read length of 4805 bp. Using a custom read depth-based analysis, we successfully confirmed the presence of all 55 known CNVs (including replicates) and the absence of one false-positive CNV. Using the same CNV-targeted data, we compared genotypes of single nucleotide variant loci to verify that no sample mix-ups occurred between assays. For one case, we also used methylation detection and phasing to investigate the parental origin of a 15q11.2-q13 duplication with implications for clinical prognosis.
We present an assay that efficiently targets genomic regions to confirm clinically relevant CNVs with a concordance rate of 100%. Furthermore, we demonstrate how integration of genotype, methylation, and phasing data from the Nanopore sequencing platform can potentially simplify and shorten the diagnostic odyssey.
罕见遗传病的诊断可能是一个漫长、昂贵且复杂的过程,需要进行一系列测试,以期获得可采取行动的结果。长读测序平台提供了通过单次检测来做出明确分子诊断的机会,该检测能够检测变异、描述甲基化模式、解析复杂重排并将发现分配给长程单倍型。在这里,我们通过验证用于神经发育障碍的拷贝数变异(CNV)的确认性测试,展示了 Nanopore 长读测序的临床实用性,并说明了该平台更广泛的应用,以评估具有重要临床意义的基因组特征。
我们在 Oxford Nanopore 平台上使用自适应采样对 25 个基因组 DNA 样本和 5 个来自最初使用短读测序检测到已知或假阳性拷贝数变化的患者的血液样本进行测序。在 30 个样本(总共 50 个有重复)中,我们检测了 35 个已知的独特 CNV(总共 55 个有重复)和一个假阳性 CNV,大小从 40kb 到 155Mb,使用归一化读取深度来评估疑似 CNV 的存在或不存在。
在单个 MinION 流池上对 50 个样本(包括重复)进行测序,我们实现了平均目标平均深度为 9.5X 和平均目标读取长度为 4805bp。使用基于自定义读取深度的分析,我们成功确认了所有 55 个已知 CNV(包括重复)的存在和一个假阳性 CNV 的不存在。使用相同的 CNV 靶向数据,我们比较了单核苷酸变异位点的基因型,以验证在检测之间没有发生样本混淆。对于一个案例,我们还使用甲基化检测和相位分析来研究 15q11.2-q13 重复的父母来源,该重复与临床预后有关。
我们提出了一种高效靶向基因组区域的检测方法,以确认具有 100%一致性率的临床相关 CNV。此外,我们展示了如何整合 Nanopore 测序平台的基因型、甲基化和相位数据,从而有可能简化和缩短诊断过程。