Colín María Jesús, Aguilar Manuel Ángel, Martín M Elena
Área de Química-Física, Facultad de Ciencias, Universidad de Extremadura, Avda. de Elvas, 06006 Badajoz, Spain.
Instituto de Computación Científica Avanzada (ICCAEx), Universidad de Extremadura, Avda. de Elvas, 06006 Badajoz, Spain.
ACS Omega. 2023 May 24;8(22):19939-19949. doi: 10.1021/acsomega.3c01906. eCollection 2023 Jun 6.
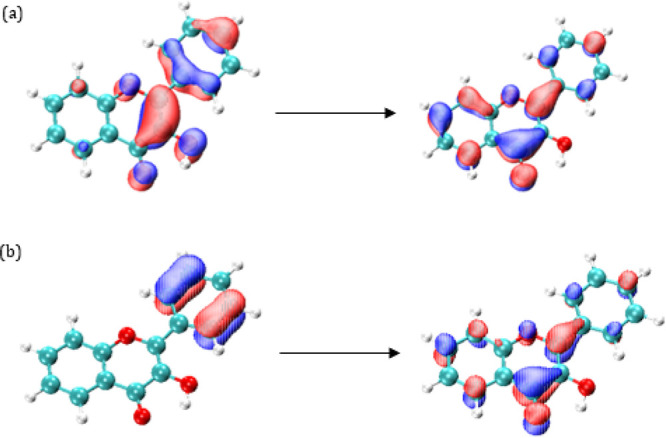
Solvent effects on the UV-vis spectra of 3-hydroxyflavone and other structurally related molecules (3-hydroxychromen-4-one, 3-hydroxy-4-pyrone, and 4-pyrone) have been studied by combining time-dependent density functional theory (TDDFT) and the polarizable continuum method (PCM). Among the first five excited states of the four considered molecules, electronic states of n → π* and π → π* nature appear. In general, the stability of the n → π* states decreases as the π space becomes larger in such a way that only for 4-pyrone and 3-hydroxy-4-pyrone are they the first excited states. In addition, they become less stabilized in ethanol solution than the ground state, and this causes blueshift transitions in solution. The opposite trend is found for the π → π* excited states. They are less energetic with the π-system size and when passing from gas phase to solution. The solvent shift also depends strongly on the size of the π systems and on the formation of an intramolecular hydrogen bond; thus, it decreases when going from 4-pyrone to 3-hydroxyflavone. The performance of the three versions (cLR, cLR, and IBSF) of the specific-state PCM method in predicting transition energies are compared.
通过结合含时密度泛函理论(TDDFT)和极化连续介质模型(PCM),研究了溶剂对3-羟基黄酮及其他结构相关分子(3-羟基色原酮-4-酮、3-羟基-4-吡喃酮和4-吡喃酮)紫外可见光谱的影响。在所研究的四个分子的前五个激发态中,出现了n→π和π→π性质的电子态。一般来说,随着π空间变大,n→π态的稳定性降低,以至于只有对于4-吡喃酮和3-羟基-4-吡喃酮,它们才是第一激发态。此外,在乙醇溶液中,它们的稳定性不如基态,这导致溶液中的跃迁发生蓝移。对于π→π激发态,情况则相反。随着π体系尺寸的增大以及从气相到溶液的变化,它们的能量降低。溶剂位移也强烈依赖于π体系的大小和分子内氢键的形成;因此,从4-吡喃酮到3-羟基黄酮时,溶剂位移减小。比较了特定状态PCM方法的三个版本(cLR、cLR和IBSF)在预测跃迁能量方面的性能。