Fili Mohammad, Hu Guiping, Han Changze, Kort Alexa, Trettin John, Haim Hillel
Department of Industrial and Manufacturing Systems Engineering, Iowa State University, 3014 Black Engineering, 2529 Union Drive, Ames, IA, 50011, USA.
Department of Microbiology and Immunology, Carver College of Medicine, University of Iowa, 51 Newton Rd, 3-770 BSB, Iowa City, IA, 52242, USA.
Algorithms Mol Biol. 2023 Jun 19;18(1):4. doi: 10.1186/s13015-023-00228-0.
Therapeutics against the envelope (Env) proteins of human immunodeficiency virus type 1 (HIV-1) effectively reduce viral loads in patients. However, due to mutations, new therapy-resistant Env variants frequently emerge. The sites of mutations on Env that appear in each patient are considered random and unpredictable. Here we developed an algorithm to estimate for each patient the mutational state of each position based on the mutational state of adjacent positions on the three-dimensional structure of the protein.
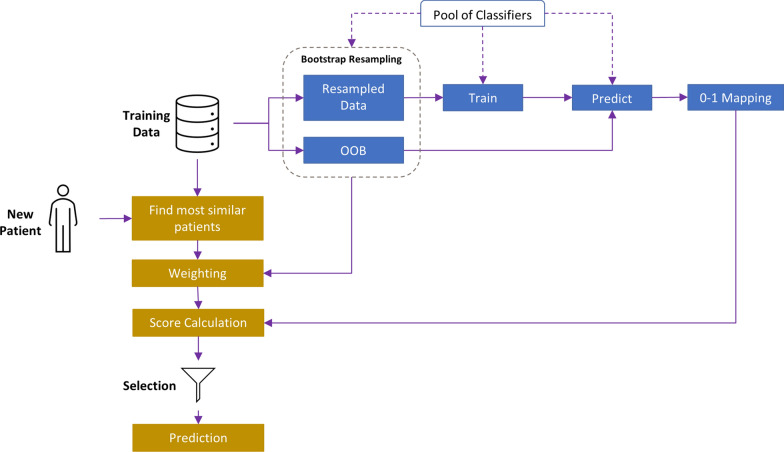
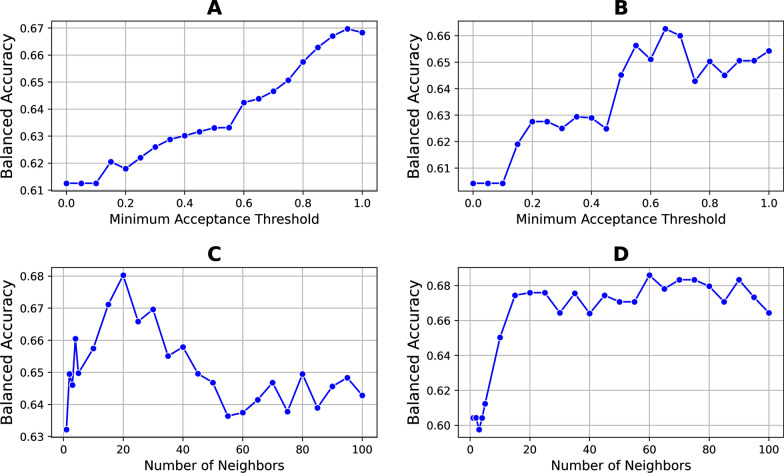
We developed a dynamic ensemble selection algorithm designated k-best classifiers. It identifies the best classifiers within the neighborhood of a new observation and applies them to predict the variability state of each observation. To evaluate the algorithm, we applied amino acid sequences of Envs from 300 HIV-1-infected individuals (at least six sequences per patient). For each patient, amino acid variability values at all Env positions were mapped onto the three-dimensional structure of the protein. Then, the variability state of each position was estimated by the variability at adjacent positions of the protein.
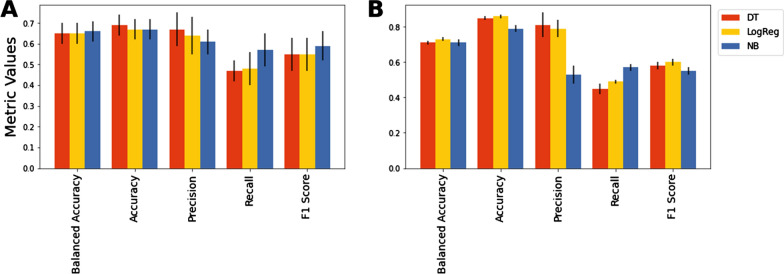
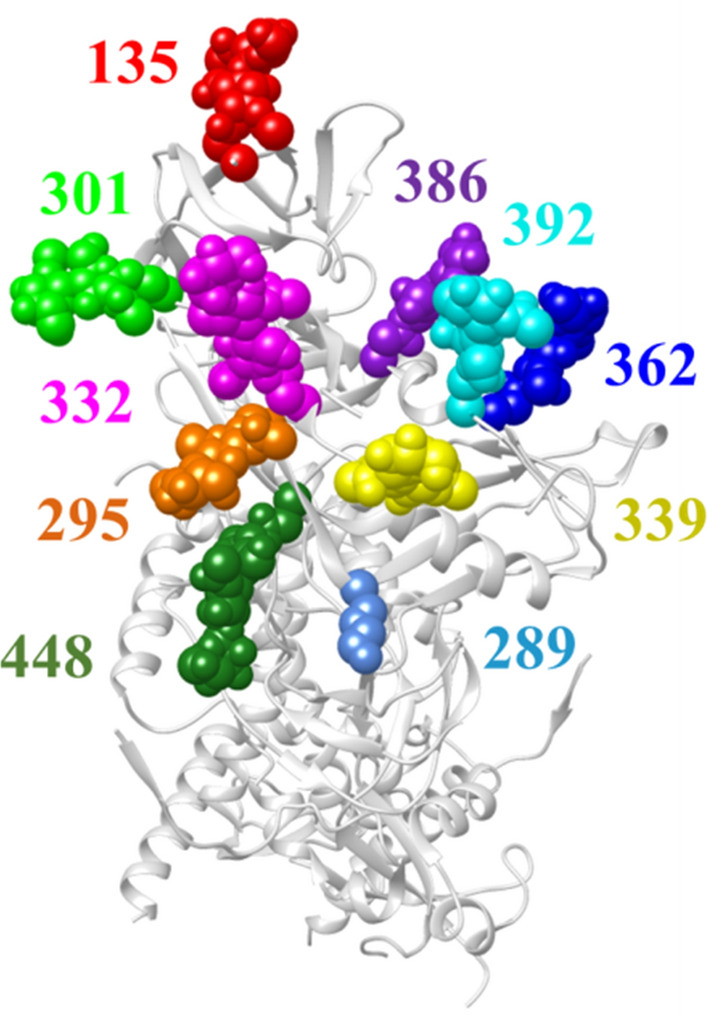
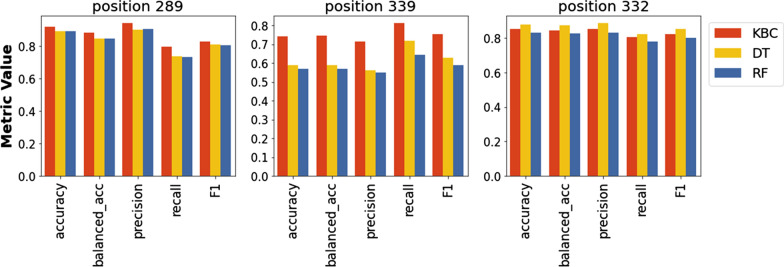
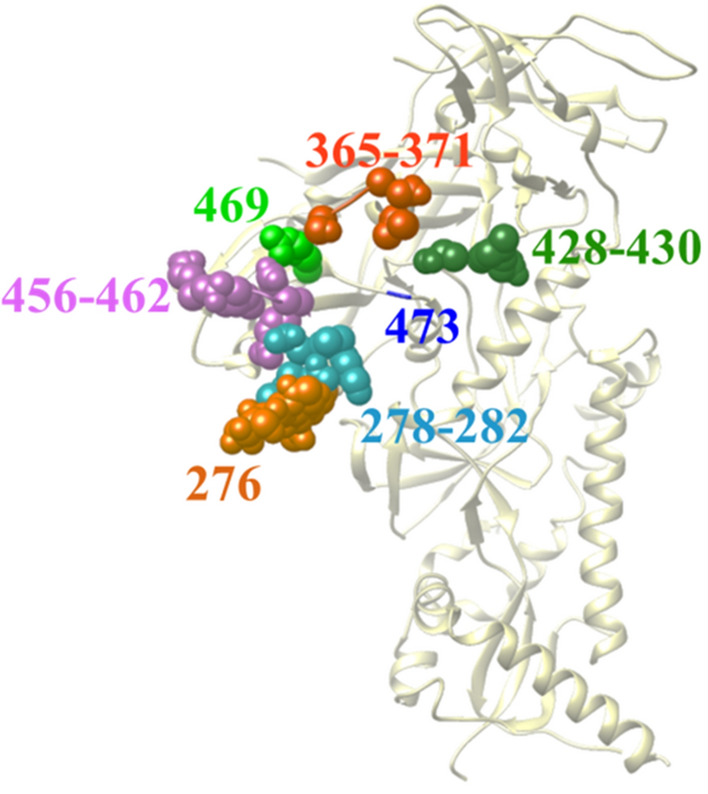
The proposed algorithm showed higher performance than the base learner and a panel of classification algorithms. The mutational state of positions in the high-mannose patch and CD4-binding site of Env, which are targeted by multiple therapeutics, was predicted well. Importantly, the algorithm outperformed other classification techniques for predicting the variability state at multi-position footprints of therapeutics on Env.
The proposed algorithm applies a dynamic classifier-scoring approach that increases its performance relative to other classification methods. Better understanding of the spatiotemporal patterns of variability across Env may lead to new treatment strategies that are tailored to the unique mutational patterns of each patient. More generally, we propose the algorithm as a new high-performance dynamic ensemble selection technique.
针对人类免疫缺陷病毒1型(HIV-1)包膜(Env)蛋白的疗法可有效降低患者体内的病毒载量。然而,由于突变,新的抗治疗Env变体频繁出现。每位患者Env上的突变位点被认为是随机且不可预测的。在此,我们开发了一种算法,可根据蛋白质三维结构上相邻位置的突变状态,为每位患者估计每个位置的突变状态。
我们开发了一种名为k-best分类器的动态集成选择算法。它在新观察值的邻域内识别最佳分类器,并将其应用于预测每个观察值的变异状态。为评估该算法,我们应用了来自300名HIV-1感染个体的Env氨基酸序列(每位患者至少六个序列)。对于每位患者,将所有Env位置的氨基酸变异值映射到蛋白质的三维结构上。然后,通过蛋白质相邻位置的变异来估计每个位置的变异状态。
所提出的算法表现出比基础学习器和一组分类算法更高的性能。Env的高甘露糖区域和CD4结合位点中多个治疗靶点位置的突变状态得到了很好的预测。重要的是,在预测治疗药物在Env上的多位点足迹处的变异状态方面,该算法优于其他分类技术。
所提出的算法应用了一种动态分类器评分方法,相对于其他分类方法提高了其性能。更好地理解Env变异的时空模式可能会带来针对每位患者独特突变模式的新治疗策略。更一般地说,我们将该算法作为一种新的高性能动态集成选择技术提出。