Neuroimmunology Unit, Montreal Neurological Institute, Department of Neurology and Neurosurgery, McGill University, 3801 Rue University, Montreal, QC, H3A 2B4, Canada.
Institute of Neuropathology, University Hospital Münster, Albert-Schweitzer-Campus 1, 48149, Münster, Germany.
Acta Neuropathol Commun. 2023 Jul 5;11(1):108. doi: 10.1186/s40478-023-01601-1.
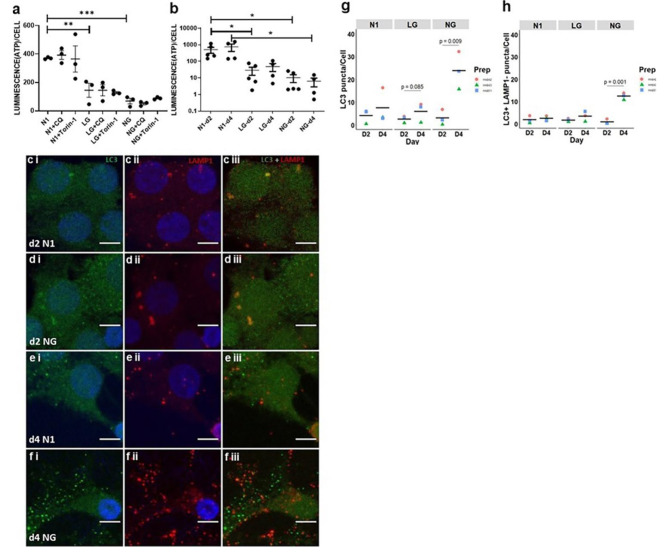
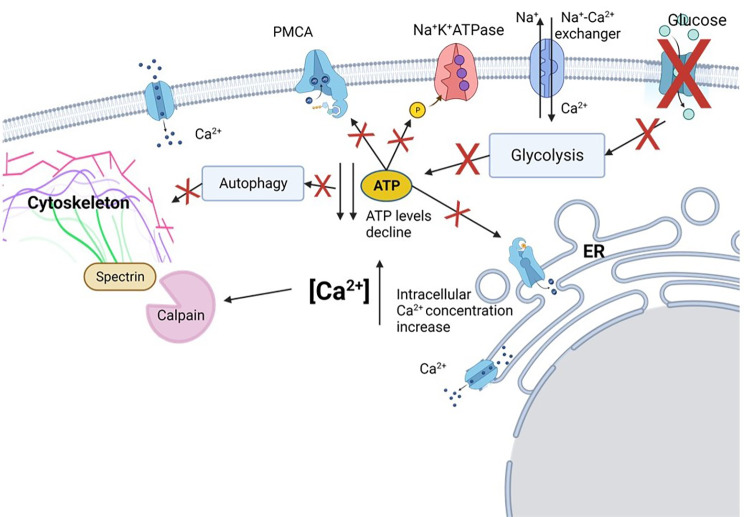
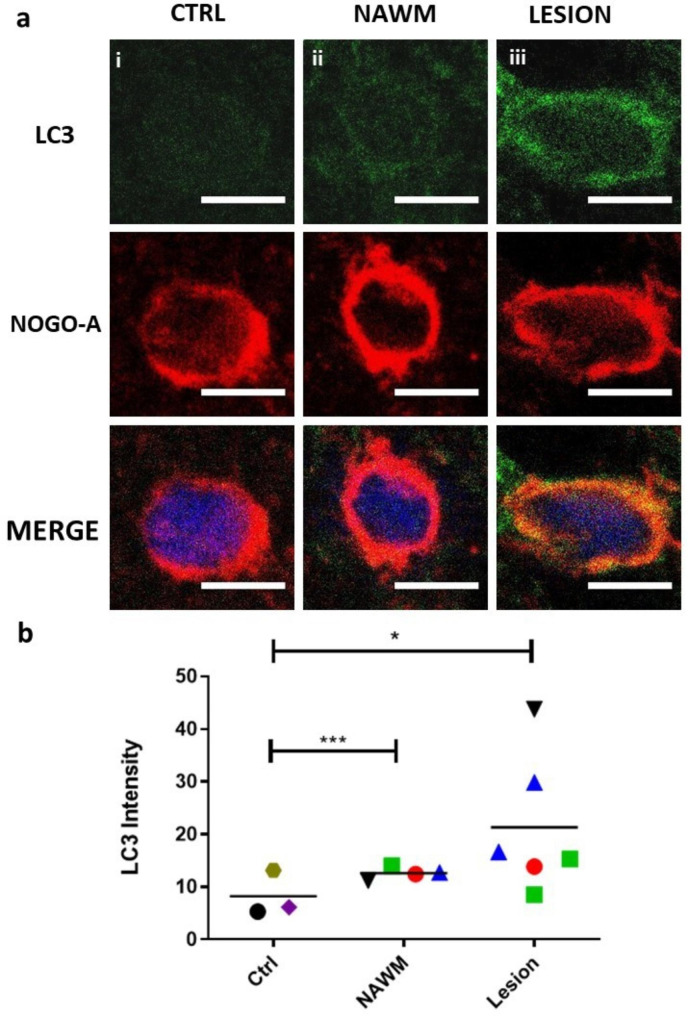
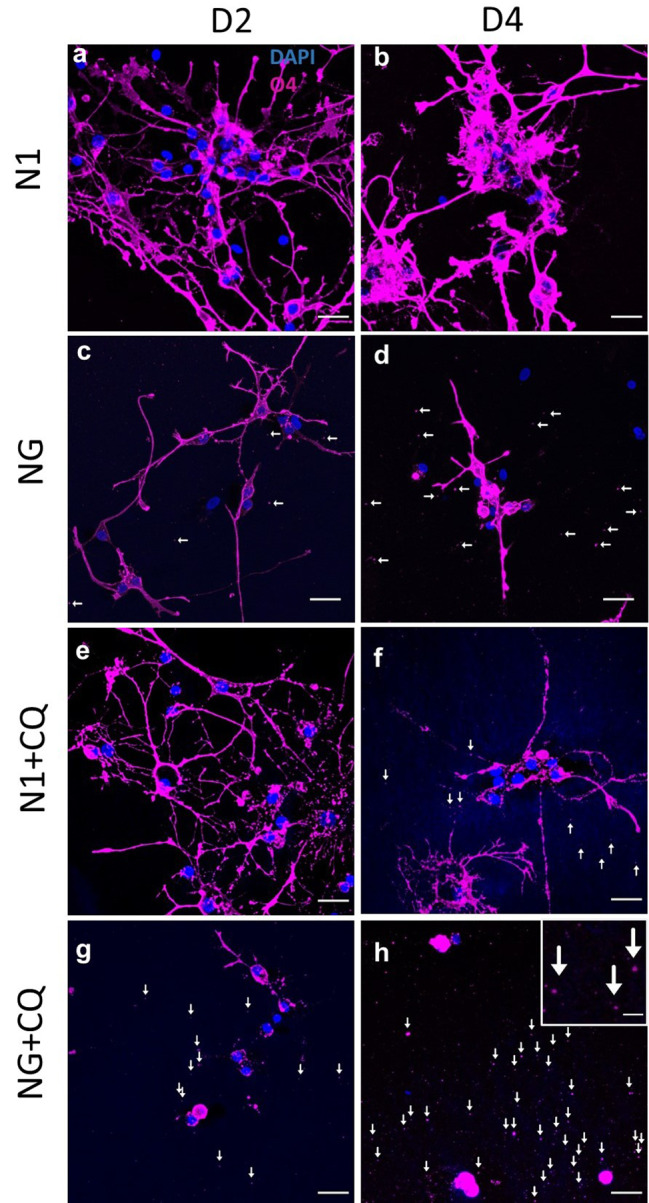
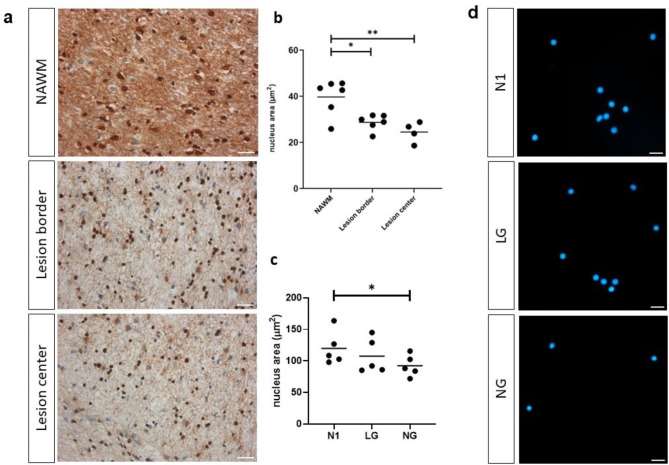
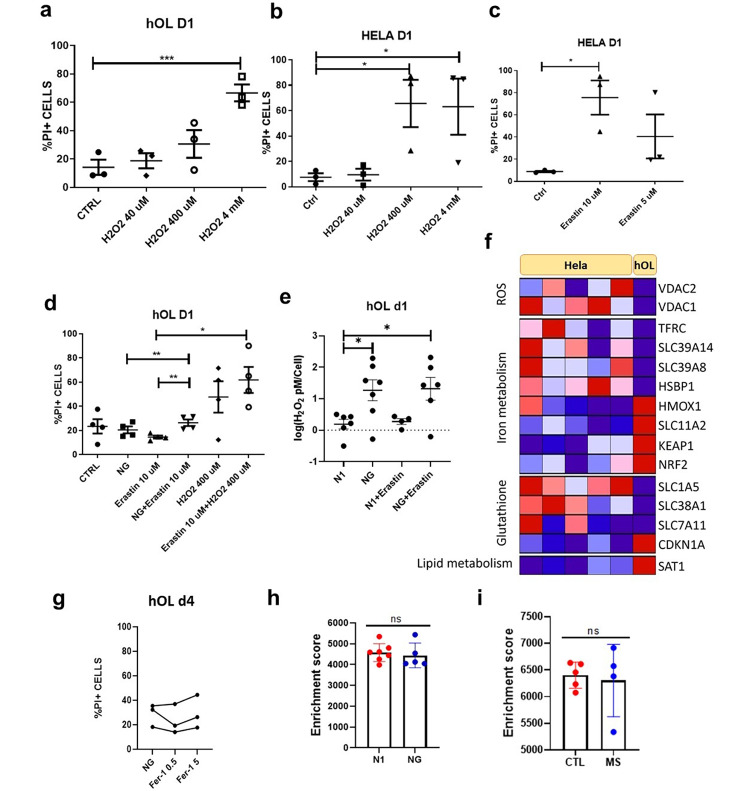
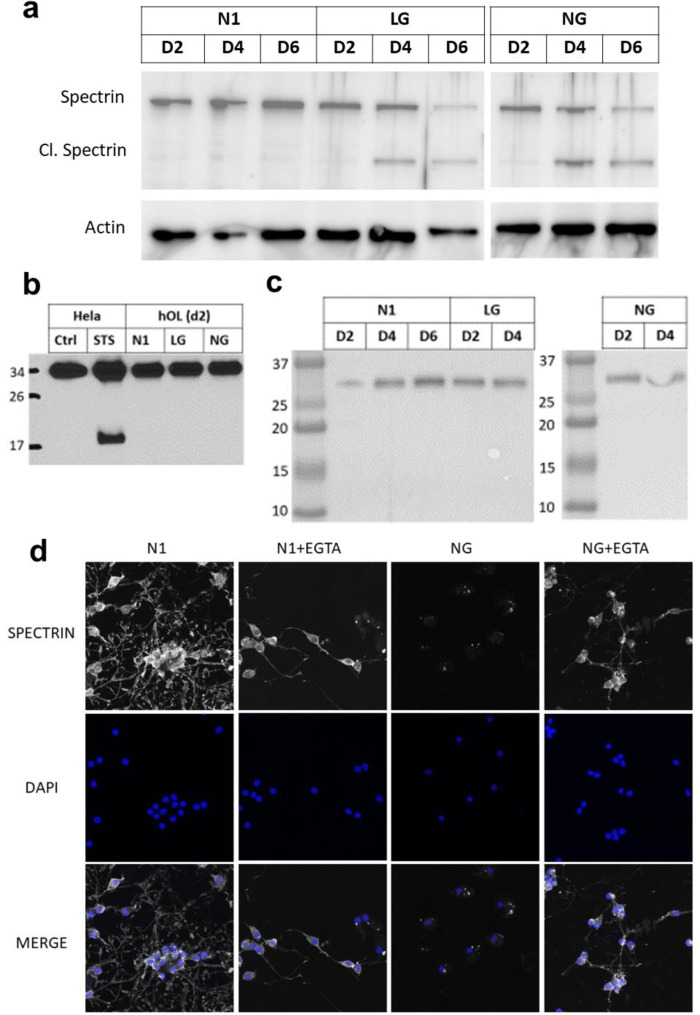
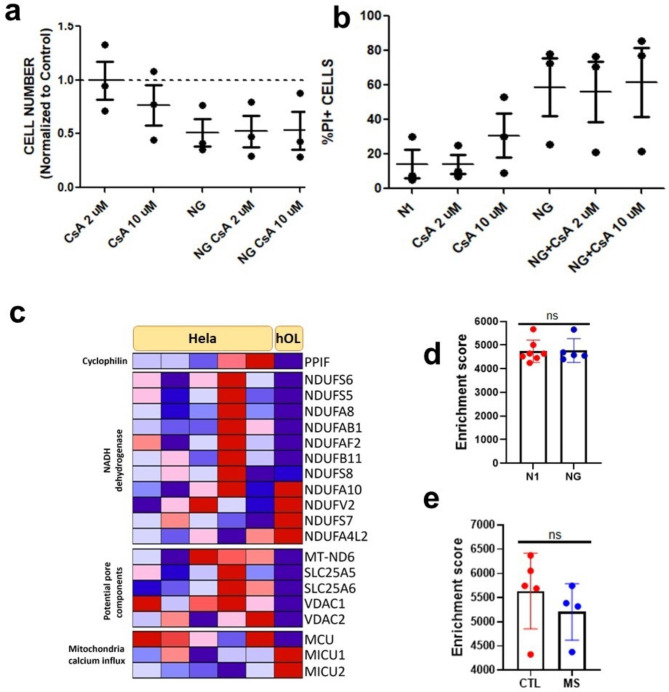
Oligodendrocyte (OL) injury and loss are central features of evolving lesions in multiple sclerosis. Potential causative mechanisms of OL loss include metabolic stress within the lesion microenvironment. Here we use the injury response of primary human OLs (hOLs) to metabolic stress (reduced glucose/nutrients) in vitro to help define the basis for the in situ features of OLs in cases of MS. Under metabolic stress in vitro, we detected reduction in ATP levels per cell that precede changes in survival. Autophagy was initially activated, although ATP levels were not altered by inhibitors (chloroquine) or activators (Torin-1). Prolonged stress resulted in autophagy failure, documented by non-fusion of autophagosomes and lysosomes. Consistent with our in vitro results, we detected higher expression of LC3, a marker of autophagosomes in OLs, in MS lesions compared to controls. Both in vitro and in situ, we observe a reduction in nuclear size of remaining OLs. Prolonged stress resulted in increased ROS and cleavage of spectrin, a target of Ca-dependent proteases. Cell death was however not prevented by inhibitors of ferroptosis or MPT-driven necrosis, the regulated cell death (RCD) pathways most likely to be activated by metabolic stress. hOLs have decreased expression of VDAC1, VDAC2, and of genes regulating iron accumulation and cyclophilin. RNA sequencing analyses did not identify activation of these RCD pathways in vitro or in MS cases. We conclude that this distinct response of hOLs, including resistance to RCD, reflects the combined impact of autophagy failure, increased ROS, and calcium influx, resulting in metabolic collapse and degeneration of cellular structural integrity. Defining the basis of OL injury and death provides guidance for development of neuro-protective strategies.
少突胶质细胞(OL)损伤和缺失是多发性硬化症进展性病变的核心特征。OL 缺失的潜在致病机制包括病变微环境中的代谢应激。在此,我们利用原代人 OL(hOL)对体外代谢应激(葡萄糖/营养物减少)的损伤反应,帮助确定 MS 病例中 OL 原位特征的基础。在体外代谢应激下,我们检测到每个细胞的 ATP 水平降低,随后细胞存活发生变化。自噬最初被激活,尽管 ATP 水平没有被抑制剂(氯喹)或激活剂(Torin-1)改变。长期应激导致自噬失败,通过自噬体和溶酶体的非融合来证明。与我们的体外结果一致,我们在 MS 病变中检测到 OL 中 LC3(自噬体的标志物)的表达升高,与对照组相比。无论是在体外还是在原位,我们都观察到剩余 OL 核大小减小。长期应激导致 ROS 增加和 spectrin 裂解,spectrin 是 Ca 依赖性蛋白酶的靶标。然而,铁死亡或 MPT 驱动的坏死抑制剂(最有可能被代谢应激激活的 RCD 途径)并没有阻止细胞死亡。hOL 的 VDAC1、VDAC2 表达降低,以及调节铁积累和细胞色素 P450 的基因表达降低。RNA 测序分析并未在体外或 MS 病例中鉴定出这些 RCD 途径的激活。我们得出结论,这种 hOL 的独特反应,包括对 RCD 的抵抗,反映了自噬失败、ROS 增加和钙内流的综合影响,导致代谢崩溃和细胞结构完整性退化。确定 OL 损伤和死亡的基础为神经保护策略的发展提供了指导。