Hort Yvonne, Sullivan Patricia, Wedd Laura, Fowles Lindsay, Stevanovski Igor, Deveson Ira, Simons Cas, Mallett Andrew, Patel Chirag, Furlong Timothy, Cowley Mark J, Shine John, Mallawaarachchi Amali
Molecular Genetics of Inherited Kidney Disorders Laboratory, Garvan Institute of Medical Research, Sydney, Australia.
Children's Cancer Institute, Lowy Cancer Centre, UNSW Sydney, Kensington, NSW, Australia.
NPJ Genom Med. 2023 Jul 7;8(1):16. doi: 10.1038/s41525-023-00362-z.
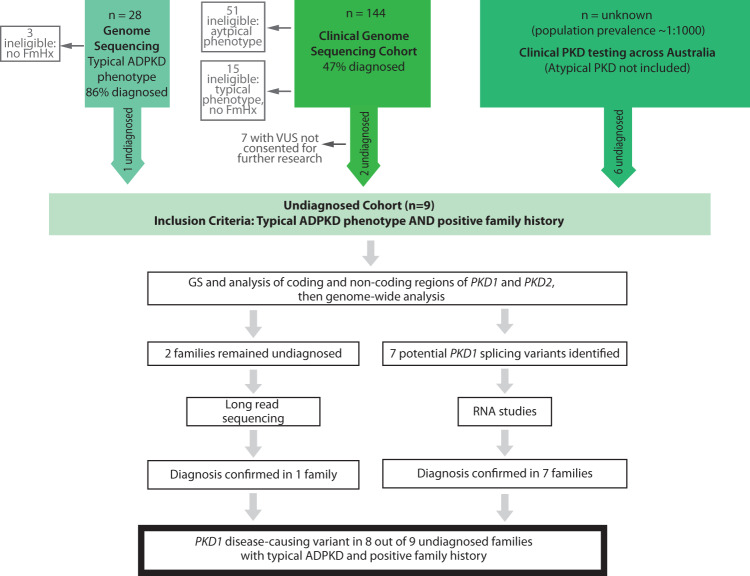
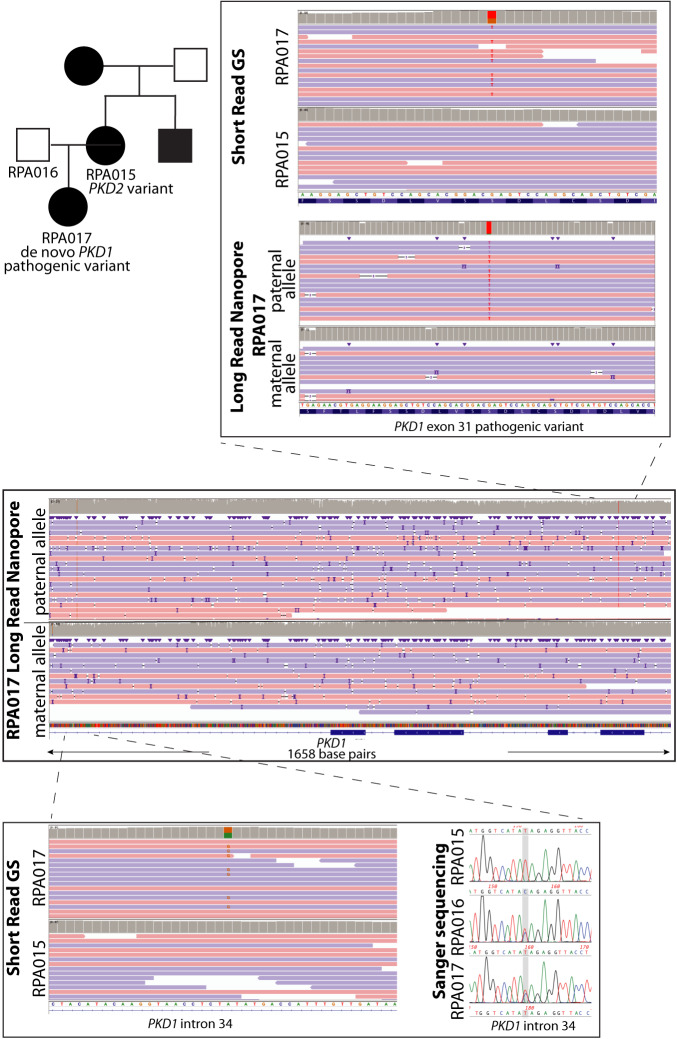
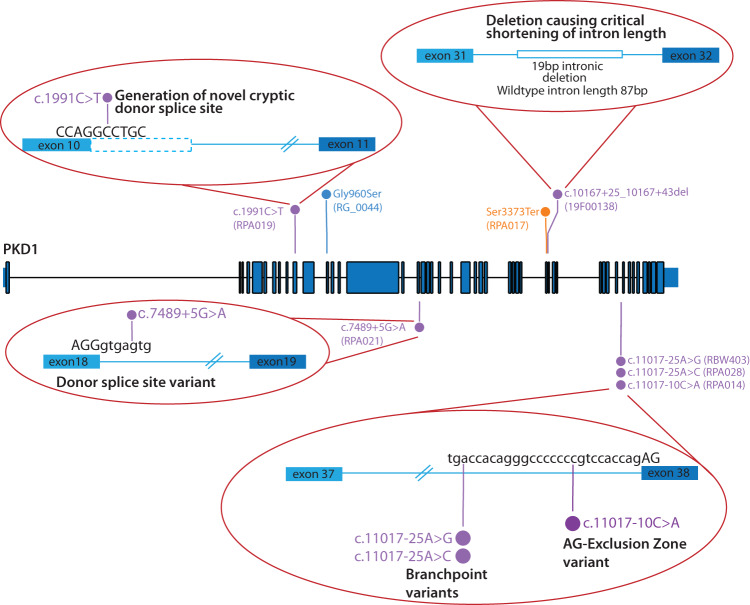
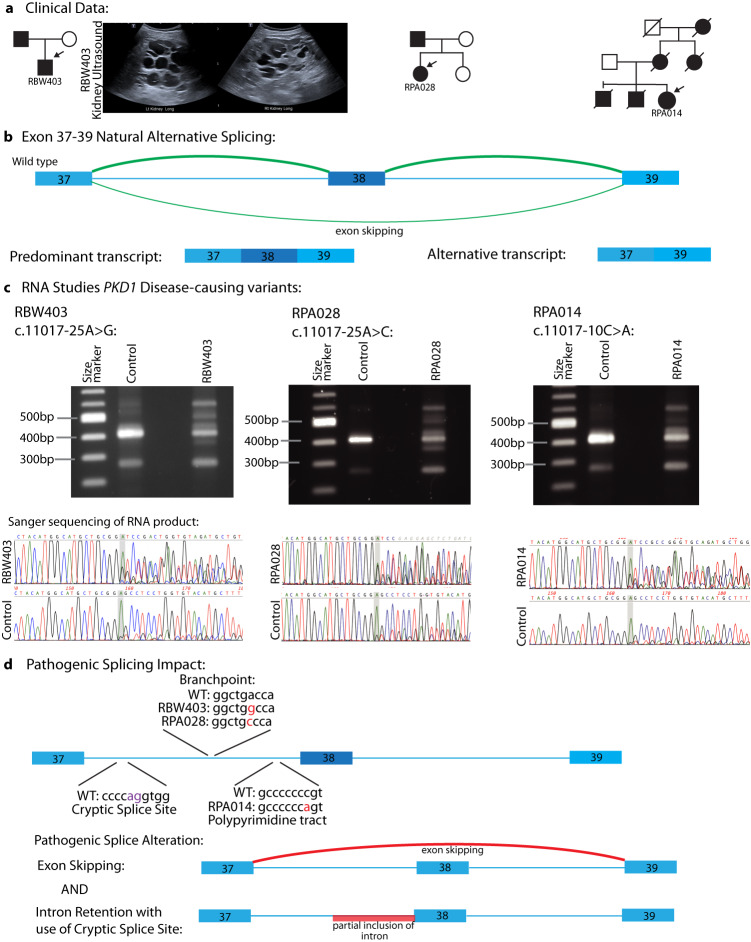
Autosomal dominant polycystic kidney disease (ADPKD) is the most common monogenic cause of kidney failure and is primarily associated with PKD1 or PKD2. Approximately 10% of patients remain undiagnosed after standard genetic testing. We aimed to utilise short and long-read genome sequencing and RNA studies to investigate undiagnosed families. Patients with typical ADPKD phenotype and undiagnosed after genetic diagnostics were recruited. Probands underwent short-read genome sequencing, PKD1 and PKD2 coding and non-coding analyses and then genome-wide analysis. Targeted RNA studies investigated variants suspected to impact splicing. Those undiagnosed then underwent Oxford Nanopore Technologies long-read genome sequencing. From over 172 probands, 9 met inclusion criteria and consented. A genetic diagnosis was made in 8 of 9 (89%) families undiagnosed on prior genetic testing. Six had variants impacting splicing, five in non-coding regions of PKD1. Short-read genome sequencing identified novel branchpoint, AG-exclusion zone and missense variants generating cryptic splice sites and a deletion causing critical intron shortening. Long-read sequencing confirmed the diagnosis in one family. Most undiagnosed families with typical ADPKD have splice-impacting variants in PKD1. We describe a pragmatic method for diagnostic laboratories to assess PKD1 and PKD2 non-coding regions and validate suspected splicing variants through targeted RNA studies.
常染色体显性多囊肾病(ADPKD)是肾衰竭最常见的单基因病因,主要与PKD1或PKD2相关。约10%的患者在标准基因检测后仍未确诊。我们旨在利用短读长和长读长基因组测序以及RNA研究来调查未确诊的家系。招募具有典型ADPKD表型且基因诊断后未确诊的患者。先证者接受短读长基因组测序、PKD1和PKD2编码及非编码分析,然后进行全基因组分析。靶向RNA研究调查怀疑影响剪接的变异。那些未确诊的患者随后接受牛津纳米孔技术长读长基因组测序。在172多名先证者中,9名符合纳入标准并同意参与。在9个之前基因检测未确诊的家系中,8个(89%)做出了基因诊断。6个家系有影响剪接的变异,5个在PKD1的非编码区。短读长基因组测序鉴定出产生隐蔽剪接位点的新分支点、AG排除区和错义变异以及导致关键内含子缩短的缺失。长读长测序在一个家系中确诊。大多数具有典型ADPKD的未确诊家系在PKD1中有影响剪接的变异。我们描述了一种实用方法,供诊断实验室评估PKD1和PKD2的非编码区,并通过靶向RNA研究验证怀疑的剪接变异。