Audano Peter A, Beck Christine R
The Jackson Laboratory for Genomic Medicine, Farmington, CT, USA.
Department of Genetics and Genome Sciences, Institute for Systems Genomics, University of Connecticut Health Center, Farmington, CT, USA.
bioRxiv. 2023 Jun 26:2023.06.25.546295. doi: 10.1101/2023.06.25.546295.
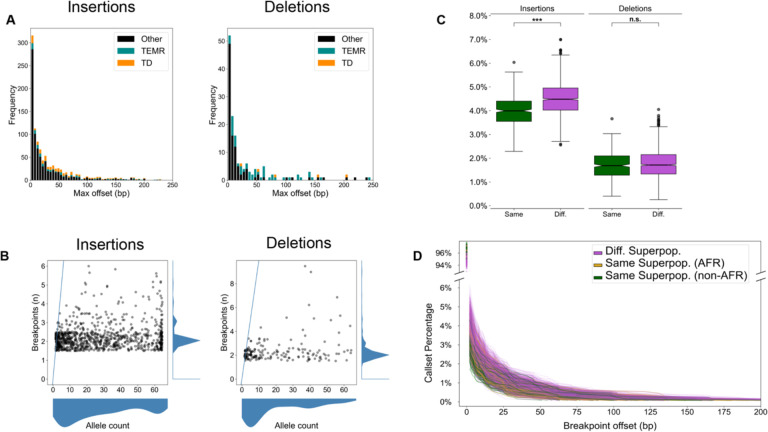
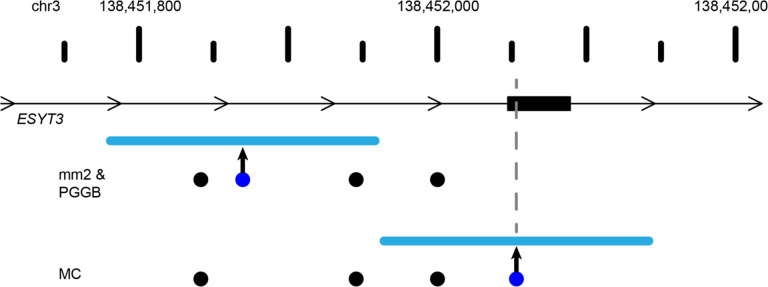
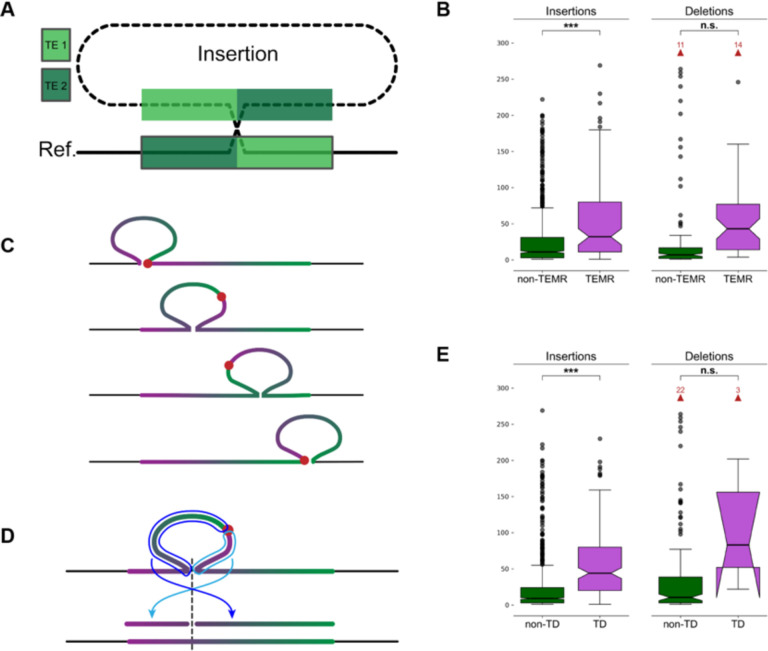
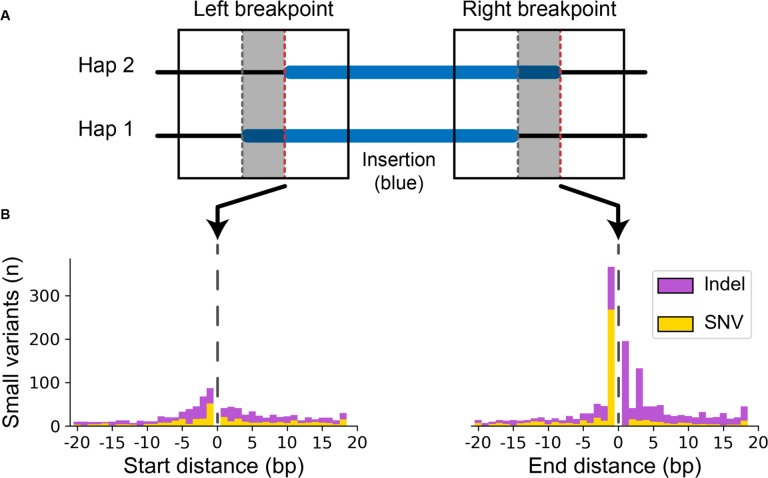
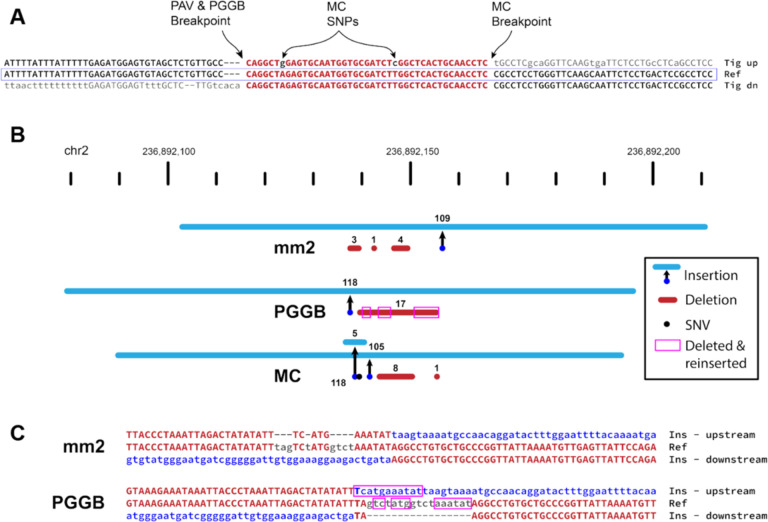
High-quality genome assemblies and sophisticated algorithms have increased sensitivity for a wide range of variant types, and breakpoint accuracy for structural variants (SVs, ≥ 50 bp) has improved to near basepair precision. Despite these advances, many SVs in unique regions of the genome are subject to systematic bias that affects breakpoint location. This ambiguity leads to less accurate variant comparisons across samples, and it obscures true breakpoint features needed for mechanistic inferences. To understand why SVs are not consistently placed, we reanalyzed 64 phased haplotypes constructed from long-read assemblies released by the Human Genome Structural Variation Consortium (HGSVC). We identified variable breakpoints for 882 SV insertions and 180 SV deletions not anchored in tandem repeats (TRs) or segmental duplications (SDs). While this is unexpectedly high for genome assemblies in unique loci, we find read-based callsets from the same sequencing data yielded 1,566 insertions and 986 deletions with inconsistent breakpoints also not anchored in TRs or SDs. When we investigated causes for breakpoint inaccuracy, we found sequence and assembly errors had minimal impact, but we observed a strong effect of ancestry. We confirmed that polymorphic mismatches and small indels are enriched at shifted breakpoints and that these polymorphisms are generally lost when breakpoints shift. Long tracts of homology, such as SVs mediated by transposable elements, increase the likelihood of imprecise SV calls and the distance they are shifted. Tandem Duplication (TD) breakpoints are the most heavily affected SV class with 14% of TDs placed at different locations across haplotypes. While graph genome methods normalize SV calls across many samples, the resulting breakpoints are sometimes incorrect, highlighting a need to tune graph methods for breakpoint accuracy. The breakpoint inconsistencies we characterize collectively affect ~5% of the SVs called in a human genome and underscore a need for algorithm development to improve SV databases, mitigate the impact of ancestry on breakpoint placement, and increase the value of callsets for investigating mutational processes.
高质量的基因组组装和复杂算法提高了对多种变异类型的检测灵敏度,结构变异(SVs,≥50 bp)的断点准确性已提高到接近碱基对的精度。尽管取得了这些进展,但基因组独特区域中的许多SVs仍存在影响断点定位的系统偏差。这种模糊性导致样本间变异比较的准确性降低,并掩盖了机制推断所需的真实断点特征。为了理解SVs定位不一致的原因,我们重新分析了由人类基因组结构变异联盟(HGSVC)发布的长读长组装构建的64个定相单倍型。我们确定了882个SV插入和180个SV缺失的可变断点,这些断点未锚定在串联重复序列(TRs)或片段重复序列(SDs)中。虽然对于独特位点的基因组组装来说,这一比例出乎意料地高,但我们发现,来自相同测序数据的基于 reads 的调用集产生了1566个插入和986个缺失,其断点也不一致,且同样未锚定在TRs或SDs中。当我们研究断点不准确的原因时,发现序列和组装错误的影响最小,但我们观察到祖先的强烈影响。我们证实,多态性错配和小插入缺失在移位断点处富集,并且当断点移位时,这些多态性通常会丢失。长片段的同源性,如由转座元件介导的SVs,增加了不精确SV调用的可能性及其移位距离。串联重复(TD)断点是受影响最严重的SV类别,14%的TDs在不同单倍型上的定位不同。虽然图形基因组方法可以对多个样本的SV调用进行标准化,但得到的断点有时是不正确的,这突出表明需要调整图形方法以提高断点准确性。我们所描述的断点不一致性共同影响了人类基因组中约5%的SVs调用,这凸显了开发算法以改进SV数据库、减轻祖先对断点定位的影响以及提高调用集对研究突变过程的价值的必要性。