Department of Pediatrics, Unit of Rare Diseases IRCCS Istituto Giannina Gaslini, Genoa, Italy.
Bambino Gesù Children'Hospital, Rome, Italy.
Orphanet J Rare Dis. 2023 Jul 21;18(1):197. doi: 10.1186/s13023-023-02797-0.
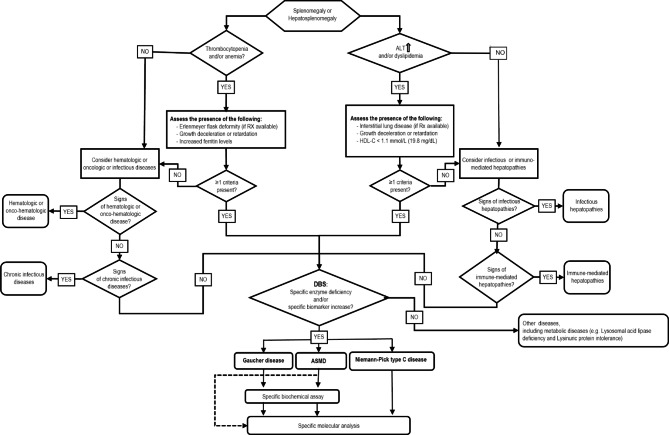
GD and ASMD are lysosomal storage disorders that enter into differential diagnosis due to the possible overlap in their clinical manifestations. The availability of safe and effective enzymatic therapies has recently led many investigators to develop and validate new screening tools, such as algorithms, for the diagnosis of LSDs where the lack of disease awareness or failure to implement newborn screening results in a delayed diagnosis.
the proposed algorithm allows for the clinical and biochemical differentiation between GD and ASMD. It is based on enzyme activity assessed on dried blood spots by multiplexed tandem mass spectrometry (MS/MS) coupled to specific biomarkers as second-tier analysis.
we believe that this method will provide a simple, convenient and sensitive tool for the screening of a selected population that can be used by pediatricians and other specialists (such as pediatric hematologists and pediatric hepatologists) often engaged in diagnosing these disorders.
GD 和 ASMD 是溶酶体贮积症,由于其临床表现可能重叠,因此需要进行鉴别诊断。由于安全有效的酶治疗方法的出现,最近许多研究人员开发并验证了新的筛选工具,例如算法,用于诊断 LSD,由于缺乏疾病意识或未能实施新生儿筛查,导致诊断延迟。
所提出的算法允许通过多维串联质谱(MS/MS)结合特定生物标志物进行的干血斑酶活性评估来对 GD 和 ASMD 进行临床和生化区分。它是作为二级分析的基于特定生物标志物的多重串联质谱法(MS/MS)。
我们认为,这种方法将为儿科医生和其他专家(例如儿科血液学家和儿科肝病学家)经常用于诊断这些疾病的选定人群提供一种简单,方便和敏感的筛选工具。