School of Intelligence Science and Technology, Key Laboratory of Machine Perception (MOE), Peking University, Beijing, 100871, China.
Department of Immunology, NHC Key Laboratory of Medical Immunology (Peking University), School of Basic Medical Sciences, Peking University Health Science Center, Beijing, China.
BMC Bioinformatics. 2023 Jul 28;24(1):302. doi: 10.1186/s12859-023-05417-7.
Single-cell RNA sequencing (scRNA-seq) enables the high-throughput profiling of gene expression at the single-cell level. However, overwhelming dropouts within data may obscure meaningful biological signals. Various imputation methods have recently been developed to address this problem. Therefore, it is important to perform a systematic evaluation of different imputation algorithms.
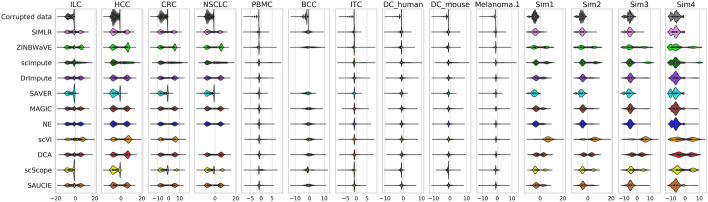
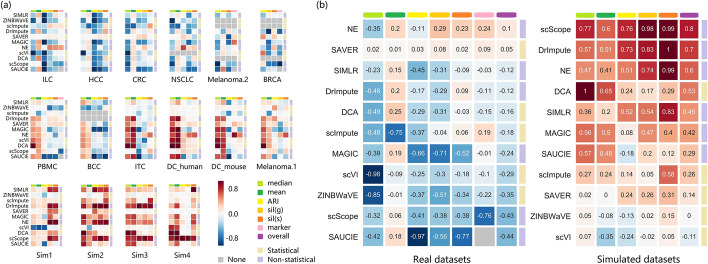
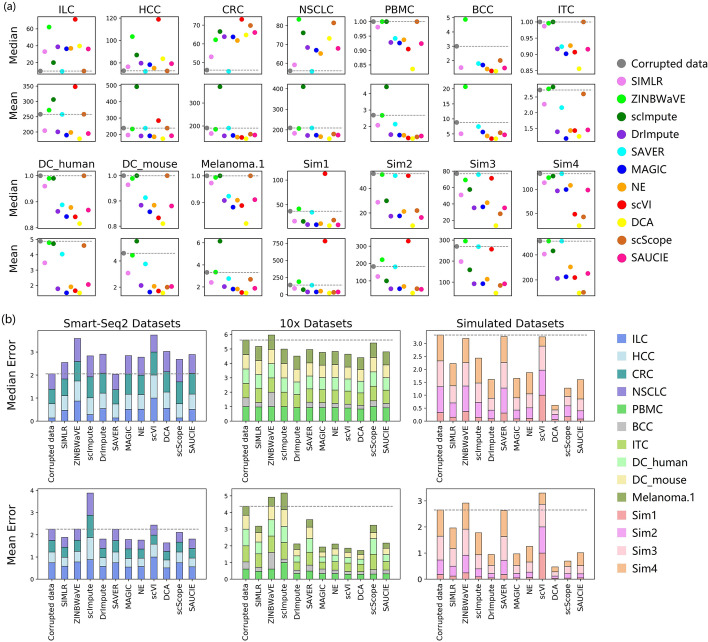
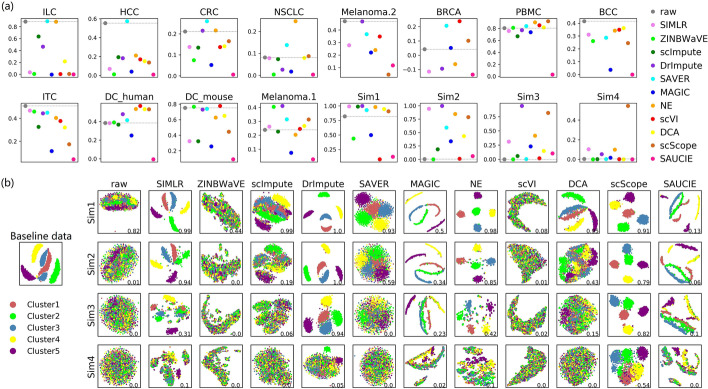
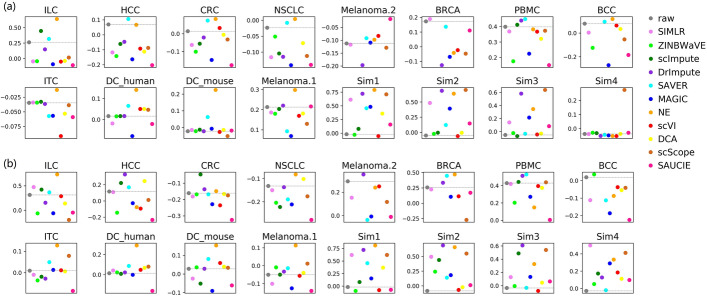
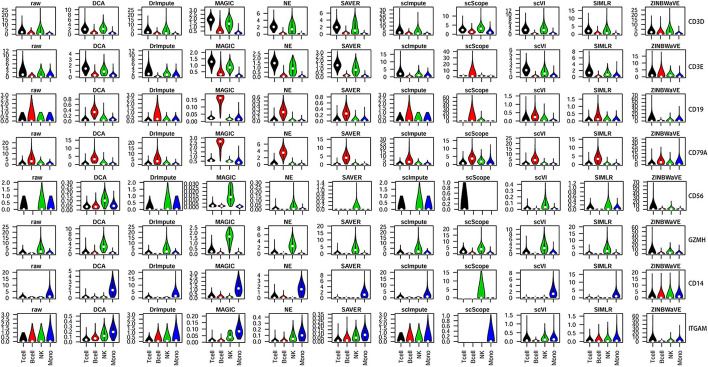
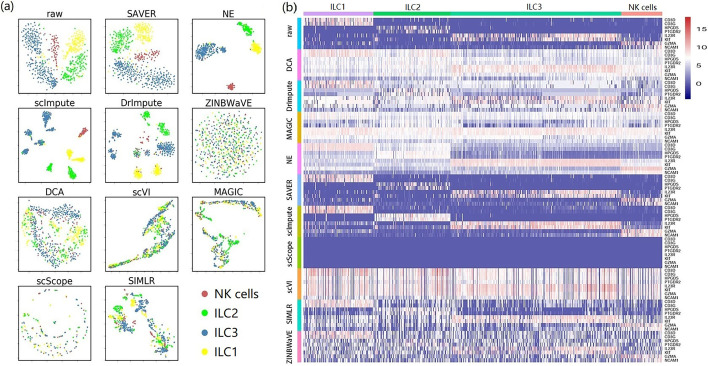
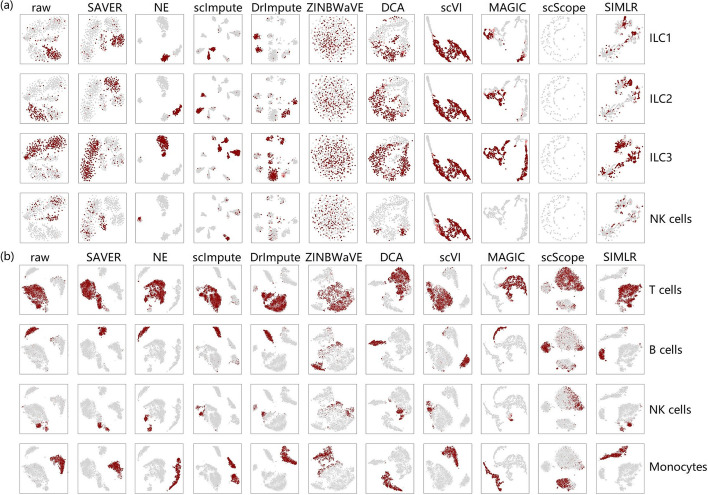
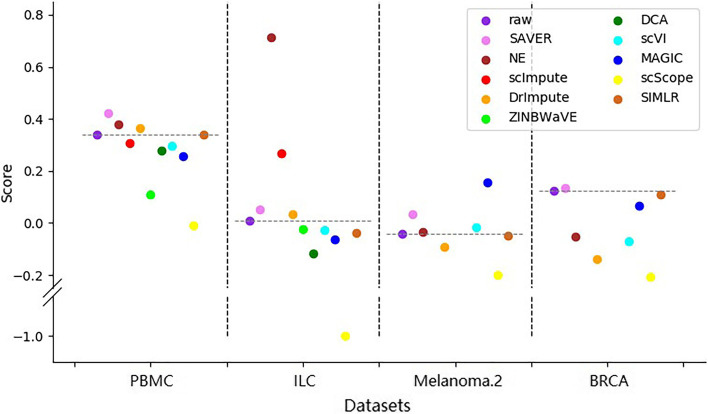
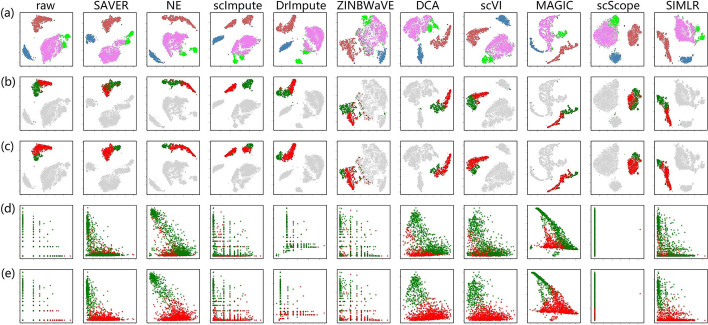
In this study, we evaluated 11 of the most recent imputation methods on 12 real biological datasets from immunological studies and 4 simulated datasets. The performance of these methods was compared, based on numerical recovery, cell clustering and marker gene analysis. Most of the methods brought some benefits on numerical recovery. To some extent, the performance of imputation methods varied among protocols. In the cell clustering analysis, no method performed consistently well across all datasets. Some methods performed poorly on real datasets but excellent on simulated datasets. Surprisingly and importantly, some methods had a negative effect on cell clustering. In marker gene analysis, some methods identified potentially novel cell subsets. However, not all of the marker genes were successfully imputed in gene expression, suggesting that imputation challenges remain.
In summary, different imputation methods showed different effects on different datasets, suggesting that imputation may have dataset specificity. Our study reveals the benefits and limitations of various imputation methods and provides a data-driven guidance for scRNA-seq data analysis.
单细胞 RNA 测序(scRNA-seq)能够在单细胞水平上高通量地分析基因表达。然而,数据中大量的缺失值可能会掩盖有意义的生物学信号。最近已经开发了各种插补方法来解决这个问题。因此,对不同的插补算法进行系统评估是很重要的。
在这项研究中,我们在 12 个来自免疫学研究的真实生物数据集和 4 个模拟数据集上评估了 11 种最新的插补方法。根据数值恢复、细胞聚类和标记基因分析,比较了这些方法的性能。大多数方法在数值恢复方面都有一定的优势。在某种程度上,插补方法的性能在不同的方案中有所不同。在细胞聚类分析中,没有一种方法在所有数据集上都表现得一致良好。一些方法在真实数据集上表现不佳,但在模拟数据集上表现出色。令人惊讶的是,一些方法对细胞聚类有负面影响。在标记基因分析中,一些方法鉴定出了潜在的新的细胞亚群。然而,并非所有的标记基因都能成功地在基因表达中进行插补,这表明插补仍然存在挑战。
总之,不同的插补方法对不同的数据集有不同的影响,这表明插补可能具有数据集特异性。我们的研究揭示了各种插补方法的优缺点,并为 scRNA-seq 数据分析提供了数据驱动的指导。