Chandra Omkar, Sharma Madhu, Pandey Neetesh, Jha Indra Prakash, Mishra Shreya, Kong Say Li, Kumar Vibhor
Department of Computational Biology, Indraprastha Institute of Information Technology, Okhla Ph-III, New Delhi, India.
Genome Institute of Singapore, Agency for Science Technology and Research, Singapore, Singapore.
Comput Struct Biotechnol J. 2023 Jul 14;21:3590-3603. doi: 10.1016/j.csbj.2023.07.014. eCollection 2023.
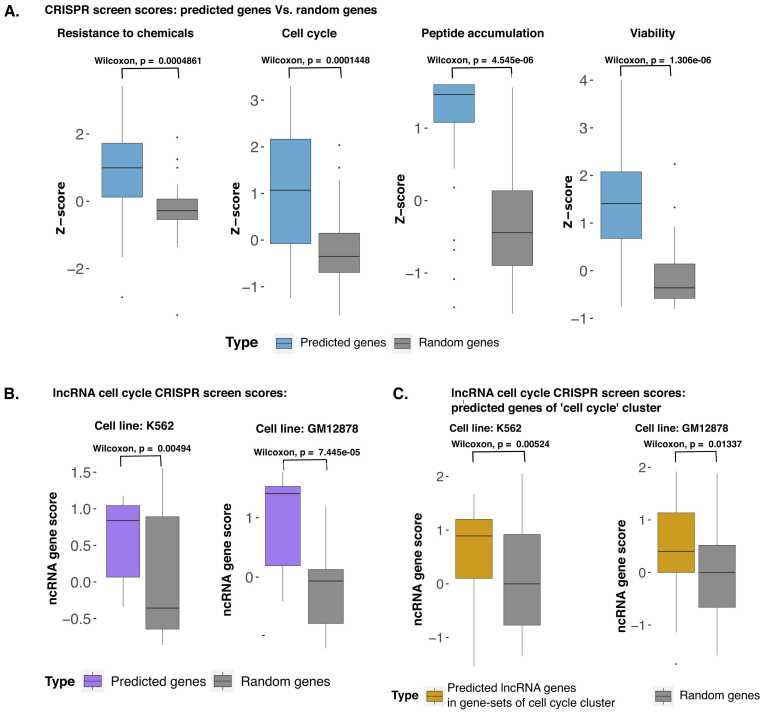
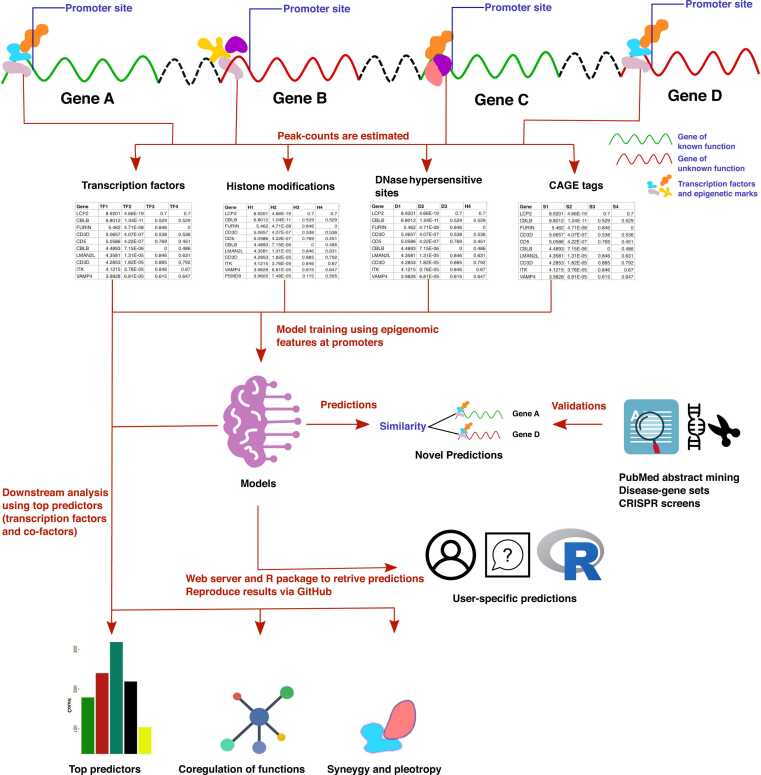
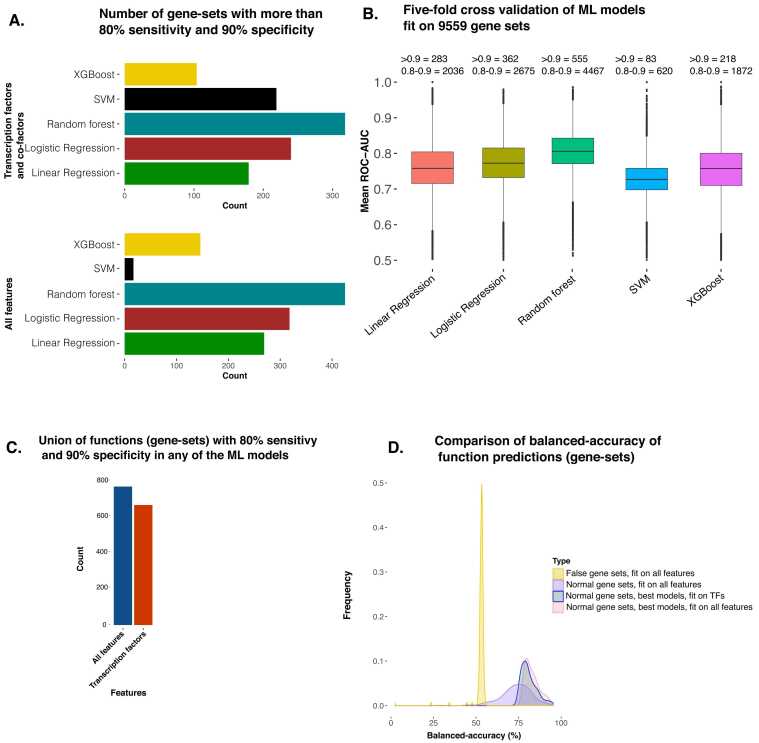
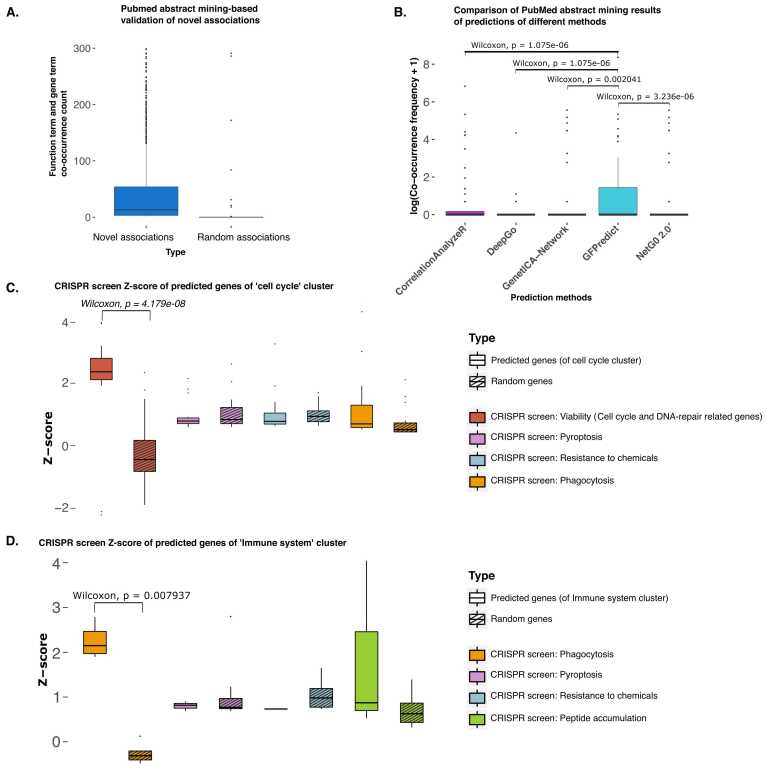
Understanding the biological roles of all genes only through experimental methods is challenging. A computational approach with reliable interpretability is needed to infer the function of genes, particularly for non-coding RNAs. We have analyzed genomic features that are present across both coding and non-coding genes like transcription factor (TF) and cofactor ChIP-seq (823), histone modifications ChIP-seq (n = 621), cap analysis gene expression (CAGE) tags (n = 255), and DNase hypersensitivity profiles (n = 255) to predict ontology-based functions of genes. Our approach for gene function prediction was reliable (>90% balanced accuracy) for 486 gene-sets. PubMed abstract mining and CRISPR screens supported the inferred association of genes with biological functions, for which our method had high accuracy. Further analysis revealed that TF-binding patterns at promoters have high predictive strength for multiple functions. TF-binding patterns at the promoter add an unexplored dimension of explainable regulatory aspects of genes and their functions. Therefore, we performed a comprehensive analysis for the functional-specificity of TF-binding patterns at promoters and used them for clustering functions to reveal many latent groups of gene-sets involved in common major cellular processes. We also showed how our approach could be used to infer the functions of non-coding genes using the CRISPR screens of coding genes, which were validated using a long non-coding RNA CRISPR screen. Thus our results demonstrated the generality of our approach by using gene-sets from CRISPR screens. Overall, our approach opens an avenue for predicting the involvement of non-coding genes in various functions.
仅通过实验方法来理解所有基因的生物学作用具有挑战性。需要一种具有可靠可解释性的计算方法来推断基因的功能,特别是对于非编码RNA。我们分析了编码基因和非编码基因共有的基因组特征,如转录因子(TF)和辅因子ChIP-seq(823个)、组蛋白修饰ChIP-seq(n = 621)、帽分析基因表达(CAGE)标签(n = 255)以及DNase超敏反应图谱(n = 255),以预测基于本体的基因功能。我们的基因功能预测方法对于486个基因集是可靠的(平衡准确率>90%)。PubMed摘要挖掘和CRISPR筛选支持了推断出的基因与生物学功能的关联,我们的方法在这方面具有很高的准确性。进一步分析表明,启动子处的TF结合模式对多种功能具有很高的预测强度。启动子处的TF结合模式为基因及其功能的可解释调控方面增添了一个未被探索的维度。因此,我们对启动子处TF结合模式的功能特异性进行了全面分析,并将其用于功能聚类,以揭示参与常见主要细胞过程的许多潜在基因集组。我们还展示了如何使用编码基因的CRISPR筛选来推断非编码基因的功能,这通过长链非编码RNA CRISPR筛选得到了验证。因此,我们的结果通过使用CRISPR筛选中的基因集证明了我们方法的通用性。总体而言,我们的方法为预测非编码基因参与各种功能开辟了一条途径。