Yoshino Ryunosuke, Yasuo Nobuaki, Hagiwara Yohsuke, Ishida Takashi, Inaoka Daniel Ken, Amano Yasushi, Tateishi Yukihiro, Ohno Kazuki, Namatame Ichiji, Niimi Tatsuya, Orita Masaya, Kita Kiyoshi, Akiyama Yutaka, Sekijima Masakazu
Transborder Medical Research Center, University of Tsukuba, Tsukuba 305-8577, Japan.
Education Academy of Computational Life Sciences (ACLS), Tokyo Institute of Technology, Yokohama 226-8501, Japan.
ACS Omega. 2023 Jul 12;8(29):25850-25860. doi: 10.1021/acsomega.3c01314. eCollection 2023 Jul 25.
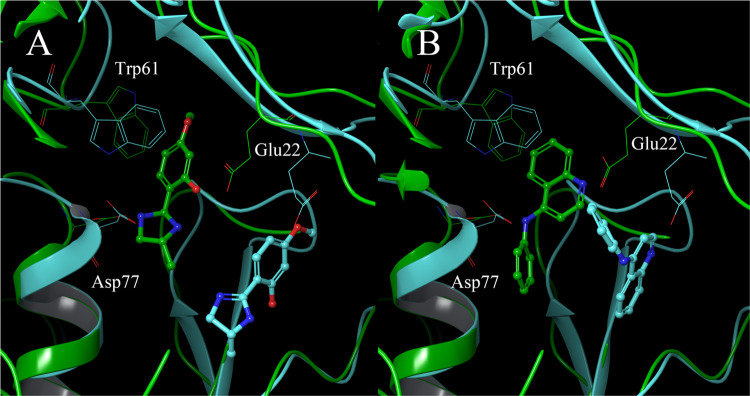
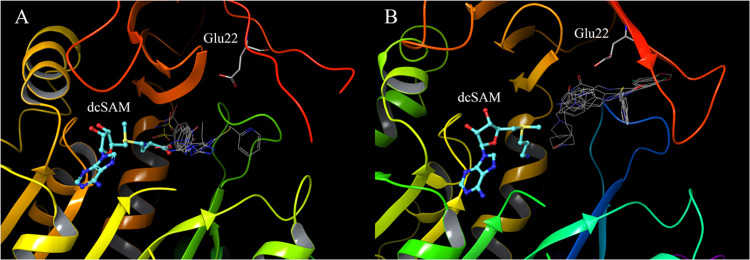
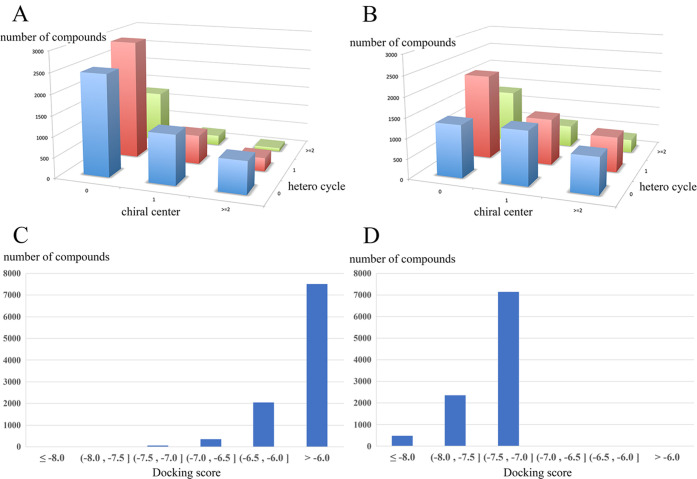
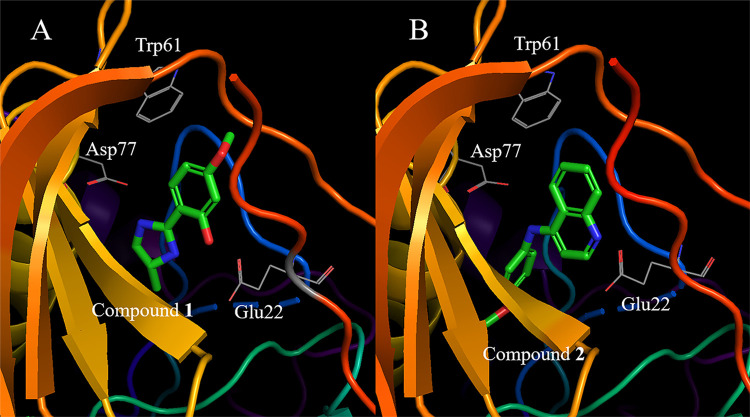
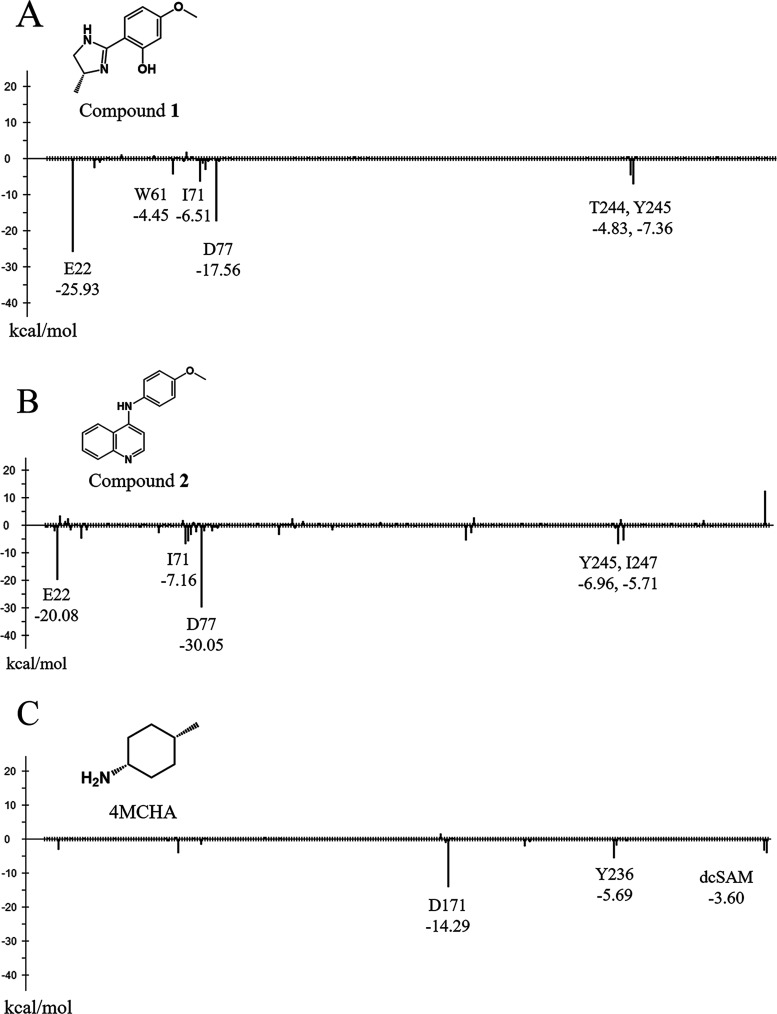
In drug discovery research, the selection of promising binding sites and understanding the binding mode of compounds are crucial fundamental studies. The current understanding of the proteins-ligand binding model extends beyond the simple lock and key model to include the induced-fit model, which alters the conformation to match the shape of the ligand, and the pre-existing equilibrium model, selectively binding structures with high binding affinity from a diverse ensemble of proteins. Although methods for detecting target protein binding sites and virtual screening techniques using docking simulation are well-established, with numerous studies reported, they only consider a very limited number of structures in the diverse ensemble of proteins, as these methods are applied to a single structure. Molecular dynamics (MD) simulation is a method for predicting protein dynamics and can detect potential ensembles of protein binding sites and hidden sites unobservable in a single-point structure. In this study, to demonstrate the utility of virtual screening with protein dynamics, MD simulations were performed on spermidine synthase to obtain an ensemble of dominant binding sites with a high probability of existence. The structure of the binding site obtained through MD simulation revealed pockets in addition to the active site that was present in the initial structure. Using the obtained binding site structures, virtual screening of 4.8 million compounds by docking simulation, assays, and X-ray analysis was conducted, successfully identifying two hit compounds.
在药物发现研究中,选择有前景的结合位点并理解化合物的结合模式是至关重要的基础研究。目前对蛋白质 - 配体结合模型的理解已超越简单的锁钥模型,包括诱导契合模型(该模型会改变构象以匹配配体形状)和预先存在的平衡模型(从多种蛋白质集合中选择性结合具有高结合亲和力的结构)。尽管检测靶蛋白结合位点的方法以及使用对接模拟的虚拟筛选技术已很成熟,并有大量研究报道,但由于这些方法应用于单一结构,所以在多种蛋白质集合中仅考虑了非常有限数量的结构。分子动力学(MD)模拟是一种预测蛋白质动力学的方法,它可以检测蛋白质结合位点的潜在集合以及在单点结构中不可见的隐藏位点。在本研究中,为了证明基于蛋白质动力学的虚拟筛选的实用性,对亚精胺合酶进行了分子动力学模拟,以获得具有高存在概率的主要结合位点集合。通过分子动力学模拟获得的结合位点结构除了初始结构中存在的活性位点外还揭示了口袋状结构。利用获得的结合位点结构,通过对接模拟、实验分析和X射线分析对480万种化合物进行了虚拟筛选,成功鉴定出两种命中化合物。