de Verneuil H, Grandchamp B, Romeo P H, Raich N, Beaumont C, Goossens M, Nicolas H, Nordmann Y
J Clin Invest. 1986 Feb;77(2):431-5. doi: 10.1172/JCI112321.
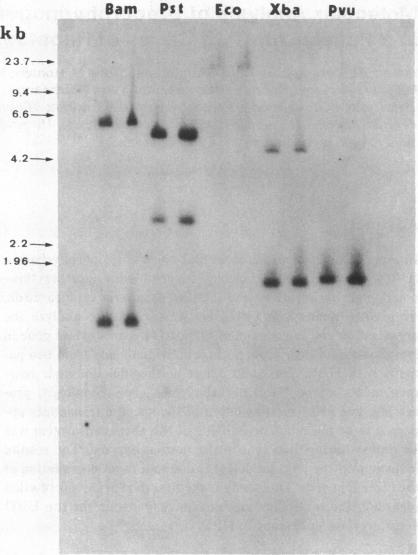
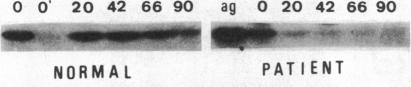
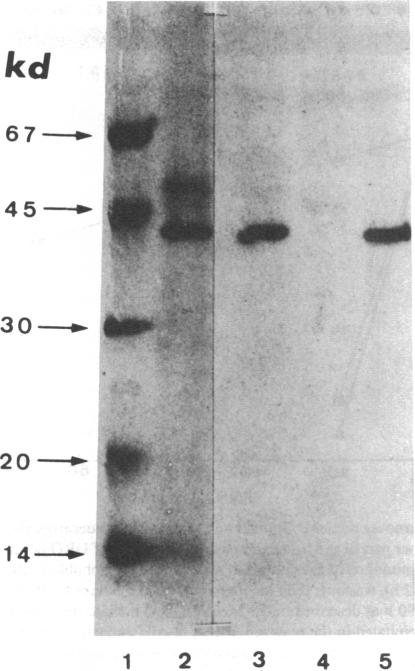
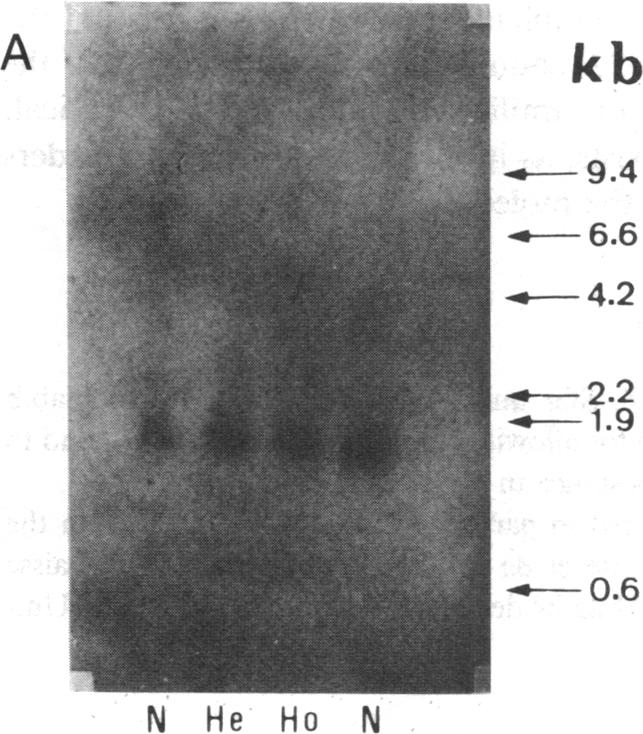
In order to determine the molecular basis of uroporphyrinogen (URO) decarboxylase deficiency responsible for hepatoerythropoietic porphyria (HEP) and familial porphyria cutanea tarda, we used a human URO decarboxylase cDNA to analyze the organization and expression of the URO decarboxylase gene in lymphoblastoid cells from normal individuals and from two patients with HEP. We could detect neither deletions nor rearrangements in the URO decarboxylase gene. Synthesis, processing, and cell-free translation of the specific transcripts appeared to be normal. The half-life of the abnormal protein was 12 times shorter than that of the normal enzyme. The results indicate that the enzyme defect is due to a rapid degradation of the protein in vivo. This study is the first to provide information regarding the molecular mechanism responsible for the URO decarboxylase deficiency in HEP.
为了确定导致肝红细胞生成性卟啉病(HEP)和家族性迟发性皮肤卟啉病的尿卟啉原(URO)脱羧酶缺乏的分子基础,我们使用人URO脱羧酶cDNA来分析正常个体以及两名HEP患者的淋巴母细胞中URO脱羧酶基因的结构和表达。我们在URO脱羧酶基因中既未检测到缺失也未检测到重排。特定转录本的合成、加工及无细胞翻译似乎均正常。异常蛋白的半衰期比正常酶的半衰期短12倍。结果表明,该酶缺陷是由于体内蛋白质的快速降解所致。本研究首次提供了有关HEP中URO脱羧酶缺乏的分子机制的信息。