Department of Pediatric, Neurology of Jilin University, 1 Xinmin Street, Changchun, 130000, Jilin Province, China.
Jilin Provincial Key Laboratory of Pediatric Neurology, Changchun, China.
BMC Med Genomics. 2023 Aug 3;16(1):181. doi: 10.1186/s12920-023-01616-6.
Pathogenic variation of the MECP2 gene presents mostly as Rett syndrome in females and is extremely rare in males. Most male patients with MECP2 gene mutation show MECP2 duplication syndrome.
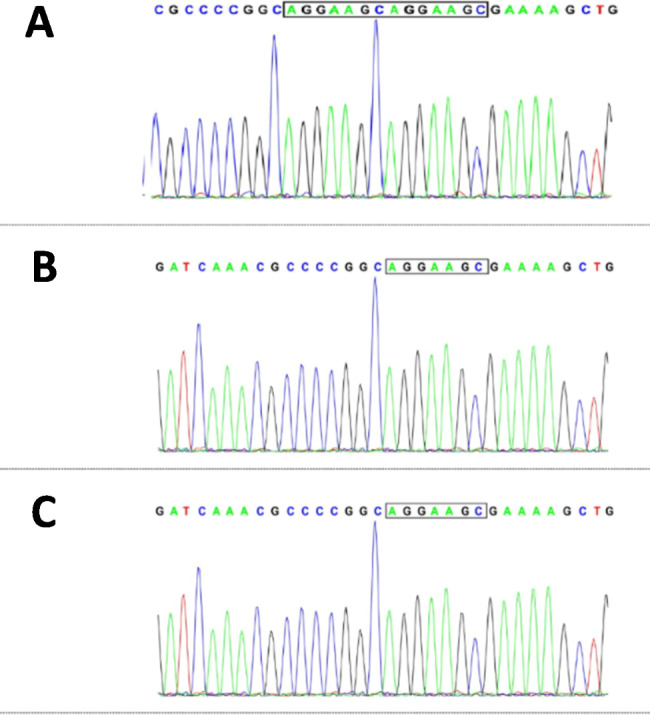
Here we report a rare case in a 10-month-old boy with a hemizygous insertion mutation in MECP2 as NM_001110792, c.799_c.800insAGGAAGC, which results in a frameshift mutation (p.R267fs*6). The patient presented with severe encephalopathy in the neonatal period, accompanied by severe development backwardness, hypotonia, and ocular and oropharyngeal dyskinesia. This is the first report of this mutation, which highlights the phenotype variability associated with MECP2 variants.
This case helps to expand the clinical spectrum associated with MECP2 variants. Close attention should be paid to the growth and development of patients carrying a MECP2 variant or Xq28 duplication. Early interventions may help improve symptoms to some certain extent.
MECP2 基因突变的致病性变异主要表现为女性的雷特综合征,在男性中极为罕见。大多数 MECP2 基因突变的男性患者表现为 MECP2 重复综合征。
本文报告了一例 10 月龄男婴罕见病例,其 MECP2 基因存在杂合插入突变 NM_001110792,c.799_c.800insAGGAAGC,导致移码突变(p.R267fs*6)。患儿在新生儿期即出现严重脑病,伴有严重发育迟缓、肌张力低下、眼球运动和口咽运动障碍。这是该突变的首次报道,突显了 MECP2 变异相关的表型多样性。
该病例有助于扩展与 MECP2 变异相关的临床谱。应密切关注携带 MECP2 变异或 Xq28 重复的患者的生长发育情况。早期干预可能有助于在一定程度上改善症状。