Faezov Bulat, Dunbrack Roland L
Institute for Cancer Research, Fox Chase Cancer Center, Philadelphia PA 19111, USA.
Kazan Federal University, Kazan, Russian Federation.
bioRxiv. 2023 Sep 3:2023.07.21.550125. doi: 10.1101/2023.07.21.550125.
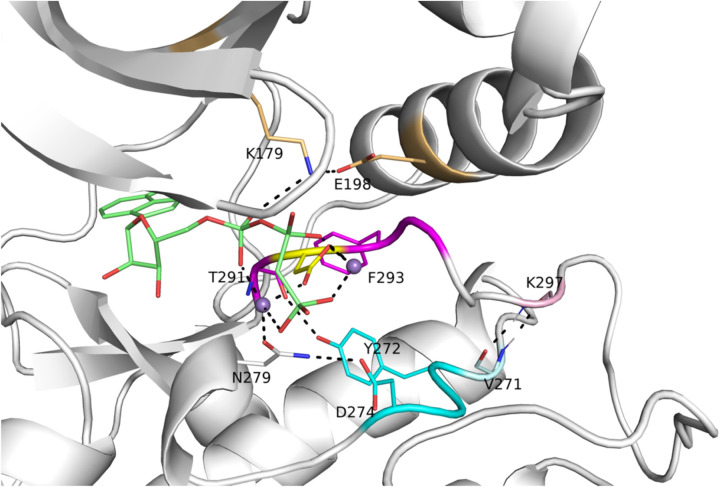
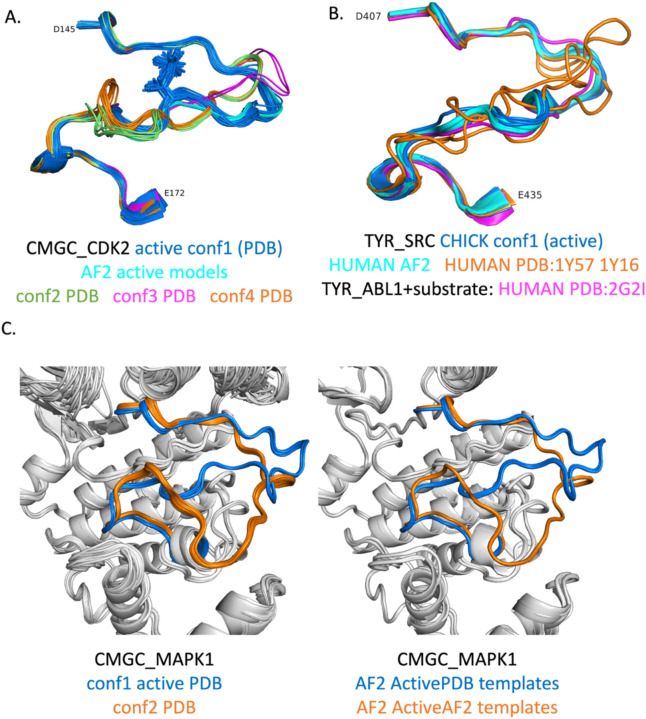
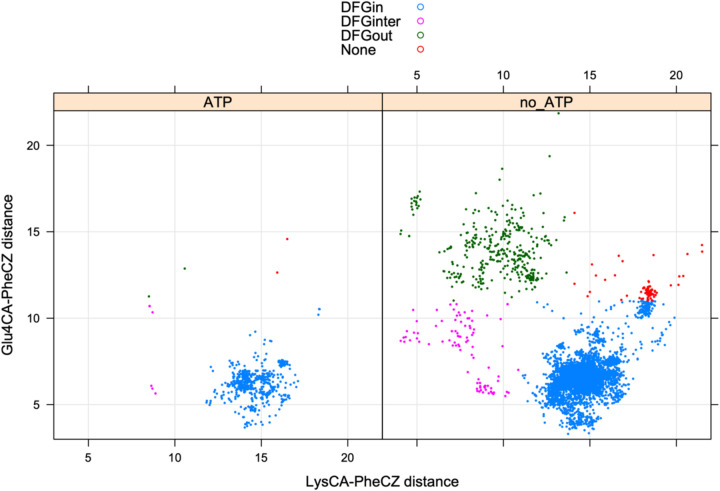
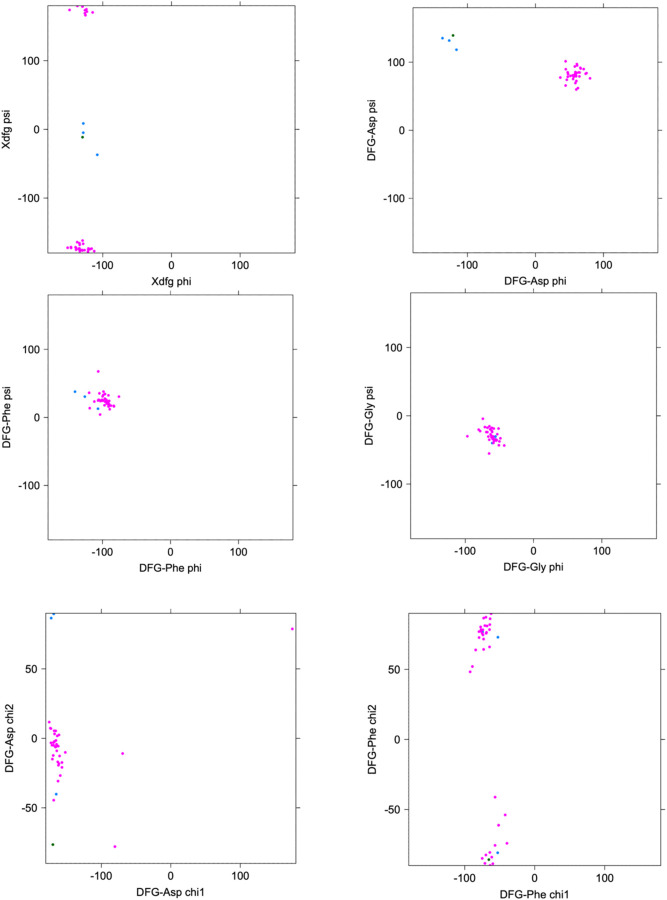
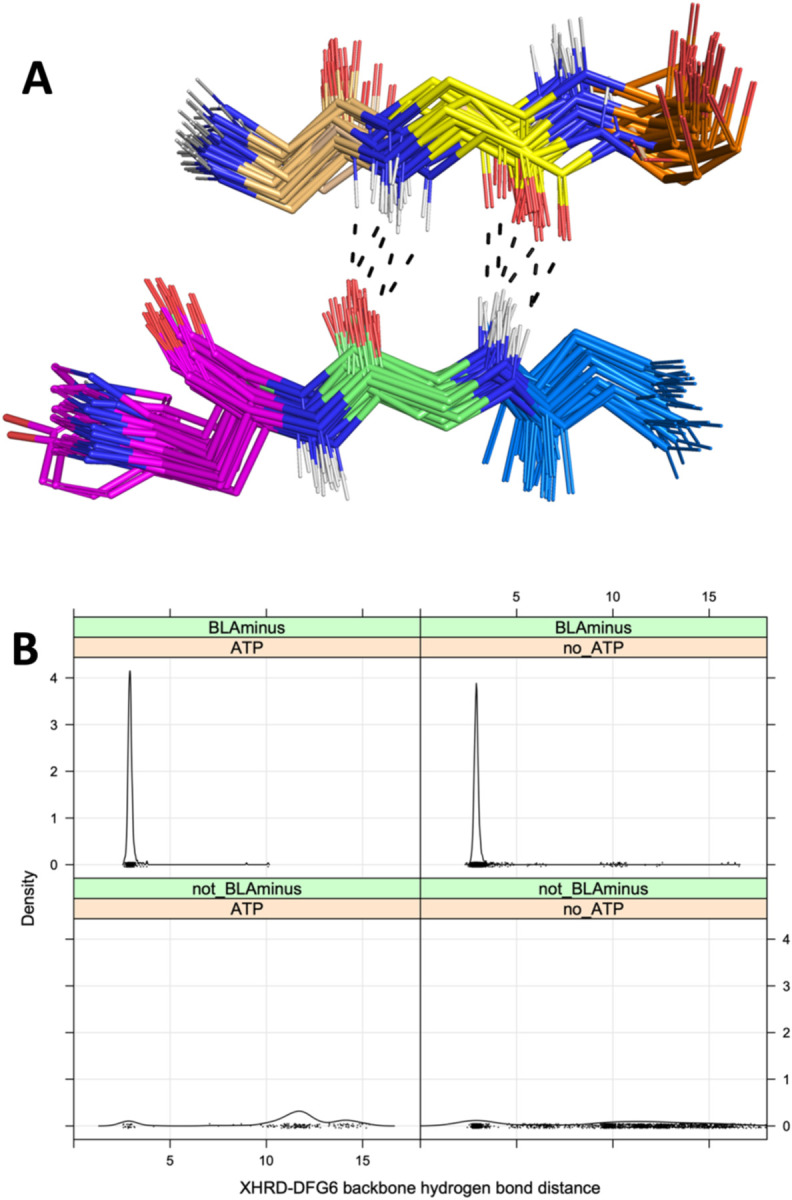
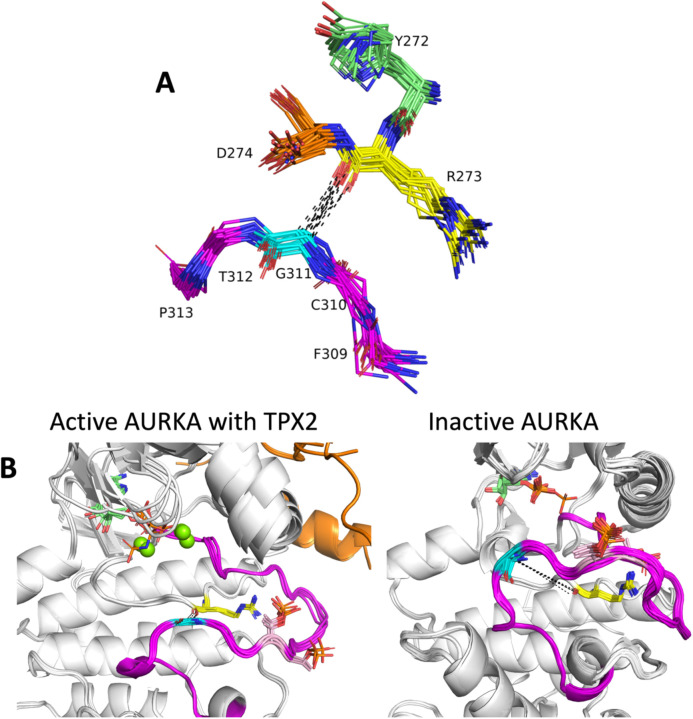
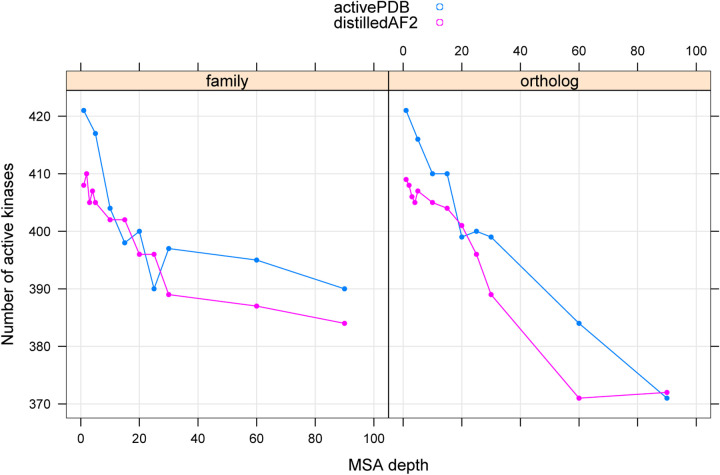
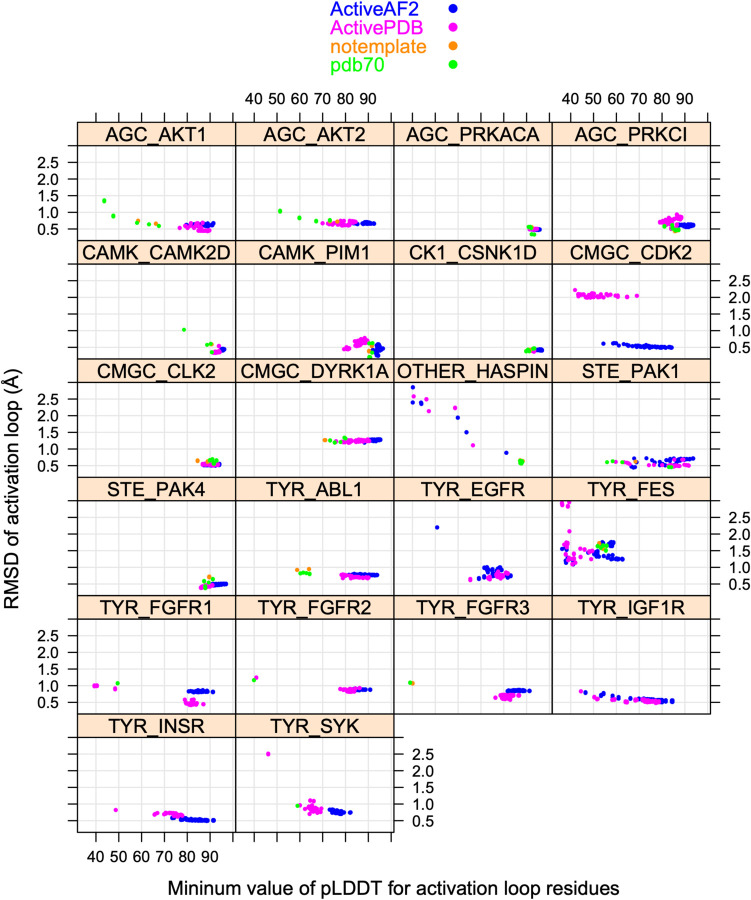
Humans have 437 catalytically competent protein kinase domains with the typical kinase fold, similar to the structure of Protein Kinase A (PKA). Only 155 of these kinases are in the Protein Data Bank in their active form. The active form of a kinase must satisfy requirements for binding ATP, magnesium, and substrate. From structural bioinformatics analysis of 40 unique substrate-bound kinases, we derived several criteria for the active form of protein kinases. We include requirements on the DFG motif of the activation loop but also on the positions of the N-terminal and C-terminal segments of the activation loop that must be placed appropriately to bind substrate. Because the active form of catalytic kinases is needed for understanding substrate specificity and the effects of mutations on catalytic activity in cancer and other diseases, we used AlphaFold2 to produce models of all 437 human protein kinases in the active form. This was accomplished with templates in the active form from the PDB and shallow multiple sequence alignments of orthologs and close homologs of the query protein. We selected models for each kinase based on the pLDDT scores of the activation loop residues, demonstrating that the highest scoring models have the lowest or close to the lowest RMSD to 22 non-redundant substrate-bound structures in the PDB. A larger benchmark of all 130 active kinase structures with complete activation loops in the PDB shows that 80% of the highest-scoring AlphaFold2 models have RMSD < 1.0 Å and 90% have RMSD < 2.0 Å over the activation loop backbone atoms. Models for all 437 catalytic kinases are available at http://dunbrack.fccc.edu/kincore/activemodels. We believe they may be useful for interpreting mutations leading to constitutive catalytic activity in cancer as well as for templates for modeling substrate and inhibitor binding for molecules which bind to the active state.
人类拥有437个具有典型激酶折叠的催化活性蛋白激酶结构域,其结构与蛋白激酶A(PKA)相似。其中只有155种激酶以活性形式存在于蛋白质数据库中。激酶的活性形式必须满足结合ATP、镁和底物的要求。通过对40种独特的底物结合激酶进行结构生物信息学分析,我们得出了蛋白质激酶活性形式的几个标准。我们不仅包括对激活环DFG基序的要求,还包括对激活环N端和C端片段位置的要求,这些片段必须适当放置以结合底物。由于理解癌症和其他疾病中底物特异性以及突变对催化活性的影响需要催化激酶的活性形式,我们使用AlphaFold2生成了所有437种人类蛋白激酶的活性形式模型。这是通过使用来自蛋白质数据库(PDB)的活性形式模板以及查询蛋白的直系同源物和近同源物的浅层多序列比对来完成的。我们根据激活环残基的pLDDT分数为每种激酶选择模型,表明得分最高的模型与蛋白质数据库中22个非冗余底物结合结构的RMSD最低或接近最低。对蛋白质数据库中所有130个具有完整激活环的活性激酶结构进行的更大规模基准测试表明,得分最高的AlphaFold2模型中80%在激活环主链原子上的RMSD < 1.0 Å,90%的RMSD < 2.0 Å。所有437种催化激酶的模型可在http://dunbrack.fccc.edu/kincore/activemodels获取。我们认为它们可能有助于解释导致癌症中组成型催化活性的突变,以及作为模拟与活性状态结合的分子的底物和抑制剂结合的模板。