Department of Chemistry, Emory University, Atlanta, Georgia 30322, United States.
Department of Pharmacology and Chemical Biology, Emory University, Atlanta, Georgia 30322, United States.
ACS Chem Neurosci. 2023 Sep 6;14(17):3059-3076. doi: 10.1021/acschemneuro.3c00181. Epub 2023 Aug 11.
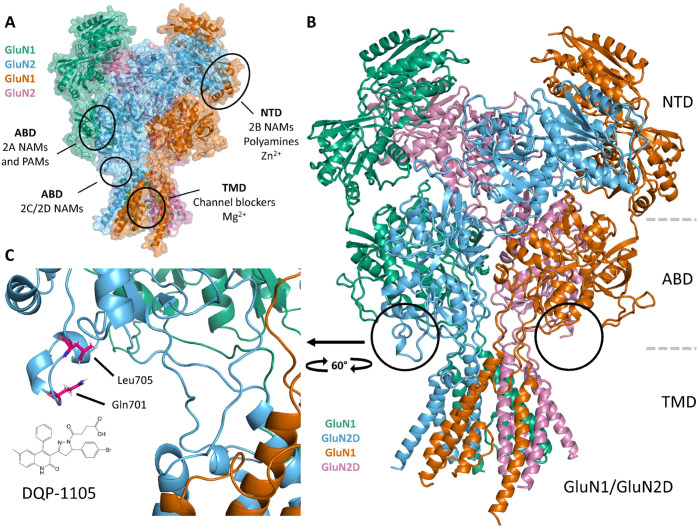
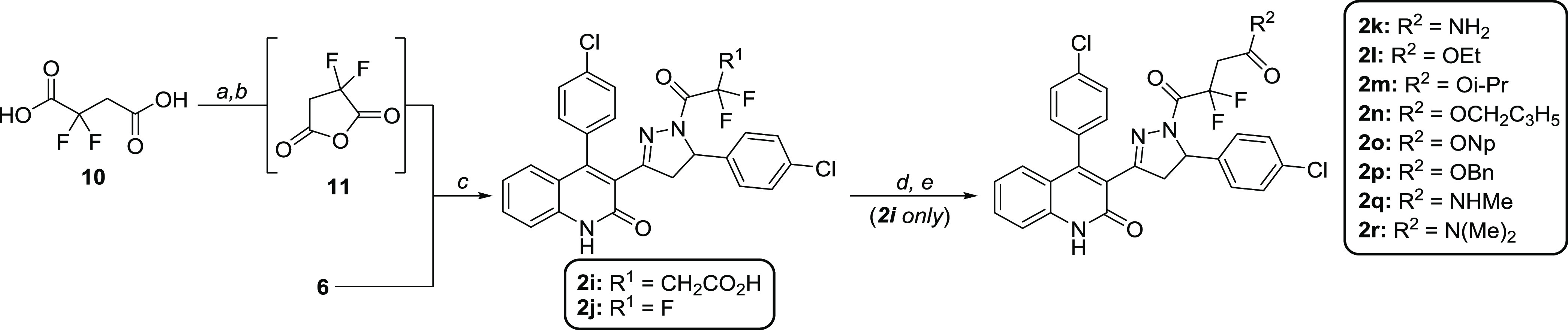
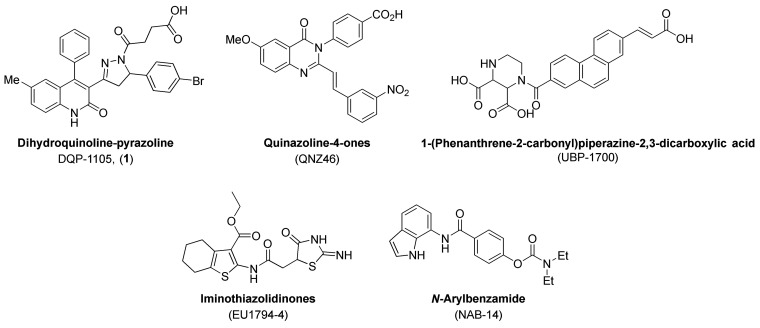
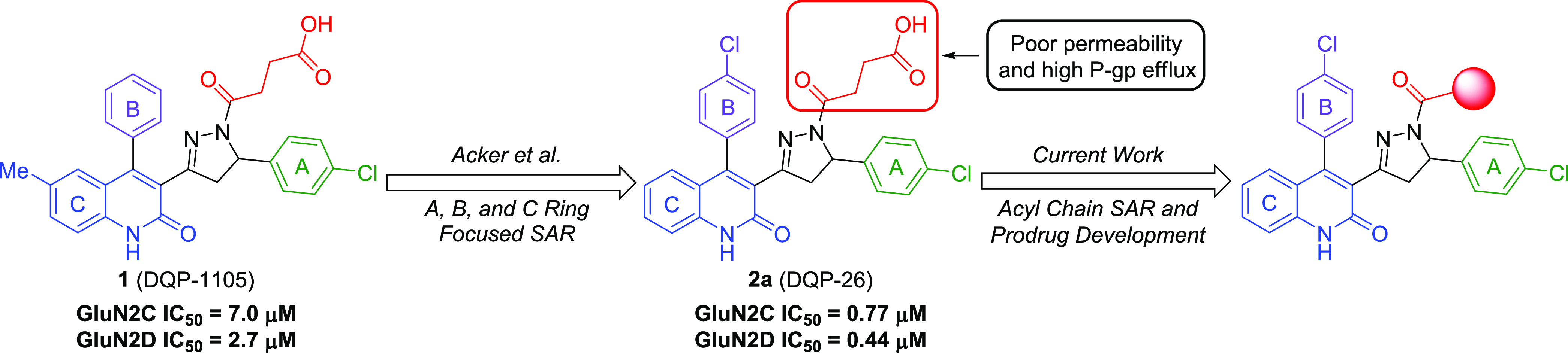
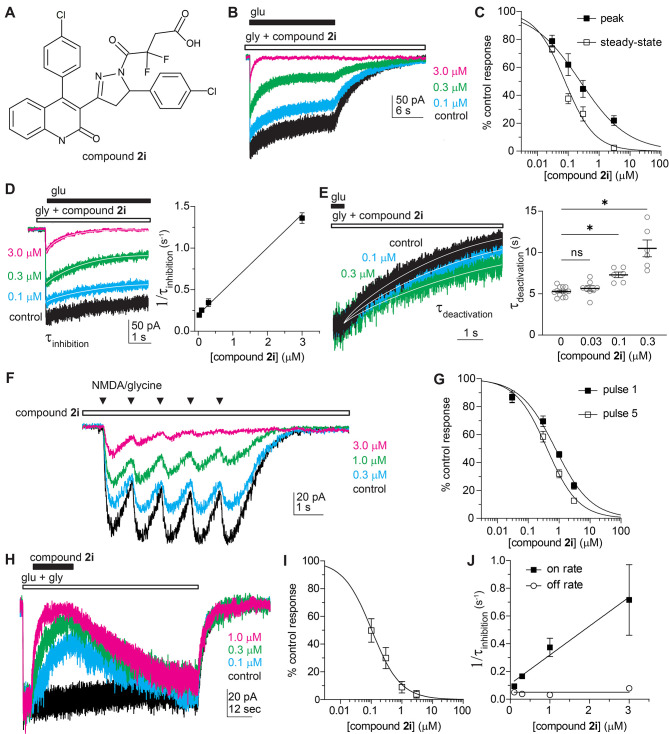
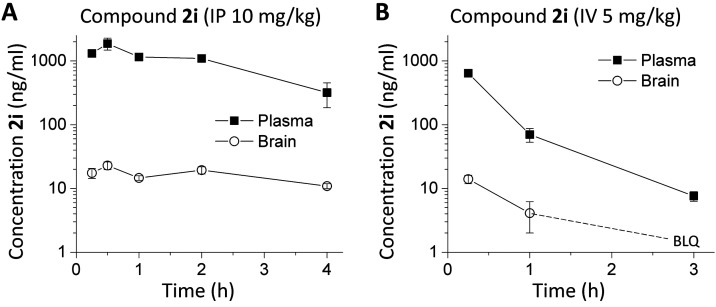
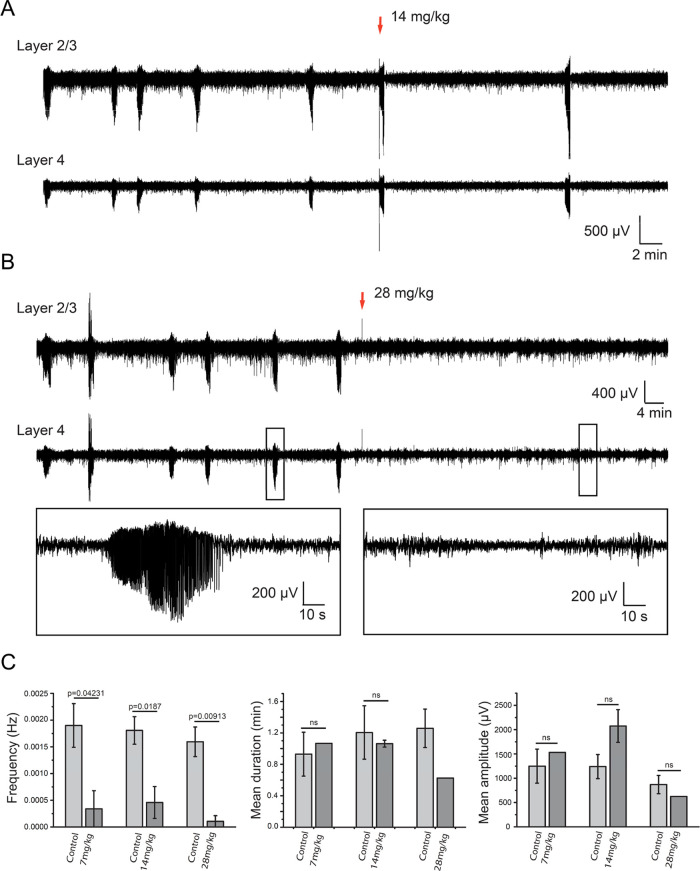
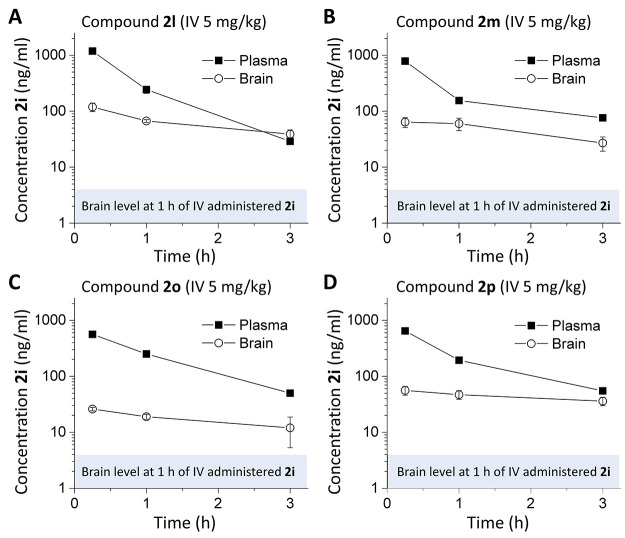
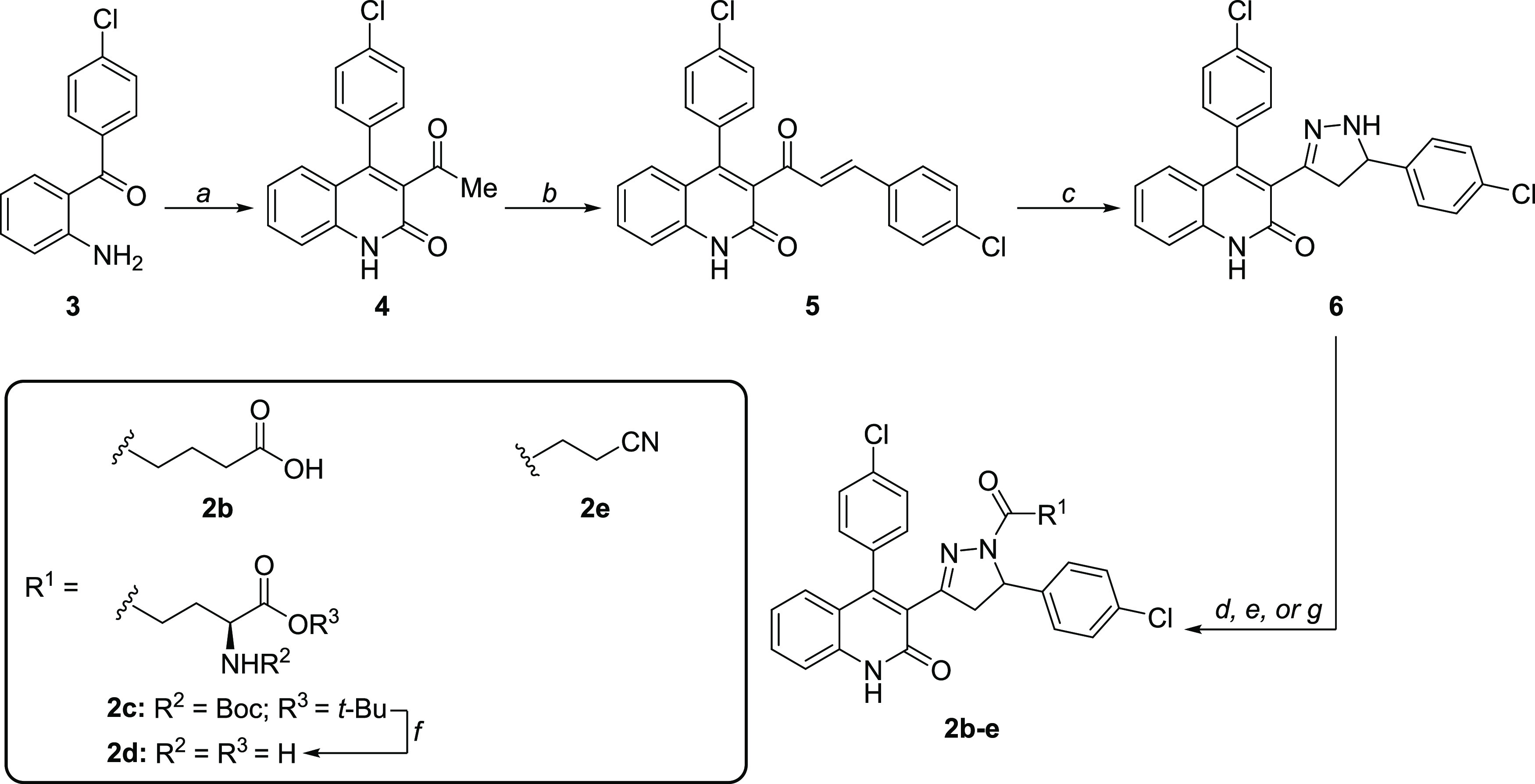
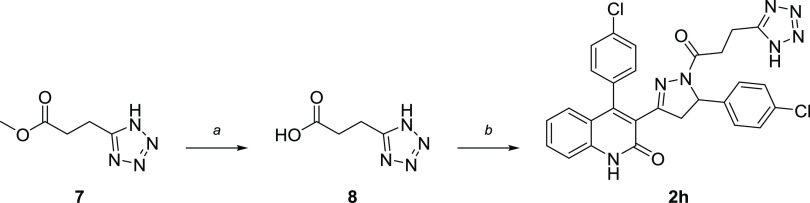
Subunit-selective inhibition of -methyl-d-aspartate receptors (NMDARs) is a promising therapeutic strategy for several neurological disorders, including epilepsy, Alzheimer's and Parkinson's disease, depression, and acute brain injury. We previously described the dihydroquinoline-pyrazoline (DQP) analogue () as a potent NMDAR negative allosteric modulator with selectivity for GluN2C/D over GluN2A/B. However, moderate (<100-fold) subunit selectivity, inadequate cell-membrane permeability, and poor brain penetration complicated the use of as an probe. In an effort to improve selectivity and the pharmacokinetic profile of the series, we performed additional structure-activity relationship studies of the succinate side chain and investigated the use of prodrugs to mask the pendant carboxylic acid. These efforts led to discovery of the analogue ()-(-)-, also referred to as ()-(-)--, which exhibits >100- and >300-fold selectivity for GluN2C- and GluN2D-containing NMDARs (IC 0.069 and 0.035 μM, respectively) compared to GluN2A- and GluN2B-containing receptors (IC 5.2 and 16 μM, respectively) and has no effects on AMPA, kainate, or GluN1/GluN3 receptors. Compound ()-(-)- is 5-fold more potent than ()-. In addition, compound shows a time-dependent enhancement of inhibitory actions at GluN2C- and GluN2D-containing NMDARs in the presence of the agonist glutamate, which could attenuate hypersynchronous activity driven by high-frequency excitatory synaptic transmission. Consistent with this finding, compound significantly reduced the number of epileptic events in a murine model of tuberous sclerosis complex (TSC)-induced epilepsy that is associated with upregulation of the GluN2C subunit. Thus, represents a robust tool for the GluN2C/D target validation. Esterification of the succinate carboxylate improved brain penetration, suggesting a strategy for therapeutic development of this series for NMDAR-associated neurological conditions.
亚基选择性抑制 -甲基-D-天冬氨酸受体 (NMDAR) 是治疗多种神经疾病的一种有前途的治疗策略,包括癫痫、阿尔茨海默病和帕金森病、抑郁症和急性脑损伤。我们之前描述了二氢喹啉-吡唑啉 (DQP) 类似物 () 作为一种有效的 NMDAR 负变构调节剂,对 GluN2C/D 具有选择性,而对 GluN2A/B 没有选择性。然而,中等 (<100 倍) 的亚基选择性、不足的细胞膜通透性和较差的脑穿透性使该化合物作为 NMDAR 的探针复杂化。为了提高该系列的选择性和药代动力学特性,我们对琥珀酸侧链进行了进一步的构效关系研究,并研究了使用前药来掩盖悬垂羧酸。这些努力导致了类似物 ()-(-)-的发现,也称为 ()-(-)--,与包含 GluN2A 和 GluN2B 的受体相比,它对包含 GluN2C 和 GluN2D 的 NMDAR 的选择性分别超过 100 倍和 300 倍(IC 0.069 和 0.035 μM,分别),而对 AMPA、 kainate 或 GluN1/GluN3 受体没有影响。化合物 ()-(-)-比 ()-更强效。此外,化合物 显示在激动剂谷氨酸存在下,对包含 GluN2C 和 GluN2D 的 NMDAR 的抑制作用具有时间依赖性增强,这可能会减轻由高频兴奋性突触传递驱动的过度同步活动。与这一发现一致的是,化合物 在与 GluN2C 亚基上调相关的结节性硬化症 (TSC) 诱导癫痫的小鼠模型中显著减少了癫痫发作的次数。因此, 代表了用于 GluN2C/D 靶标验证的强大工具。琥珀酸羧酸的酯化提高了脑穿透性,这表明了该系列用于治疗与 NMDAR 相关的神经疾病的治疗开发策略。