Choudhery Sanjeevani, DeJesus Michael A, Srinivasan Aarthi, Rock Jeremy, Schnappinger Dirk, Ioerger Thomas R
Department of Computer Science and Engineering, Texas A&M University, College Station, Texas, United States of America.
Laboratory of Host-Pathogen Biology, The Rockefeller University, New York, New York, United States of America.
bioRxiv. 2024 Feb 7:2023.08.03.551759. doi: 10.1101/2023.08.03.551759.
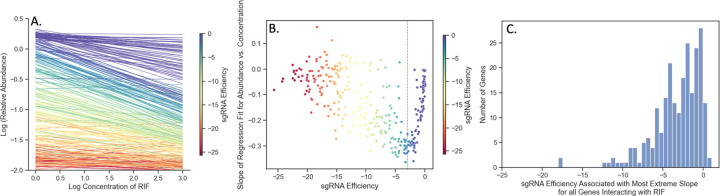
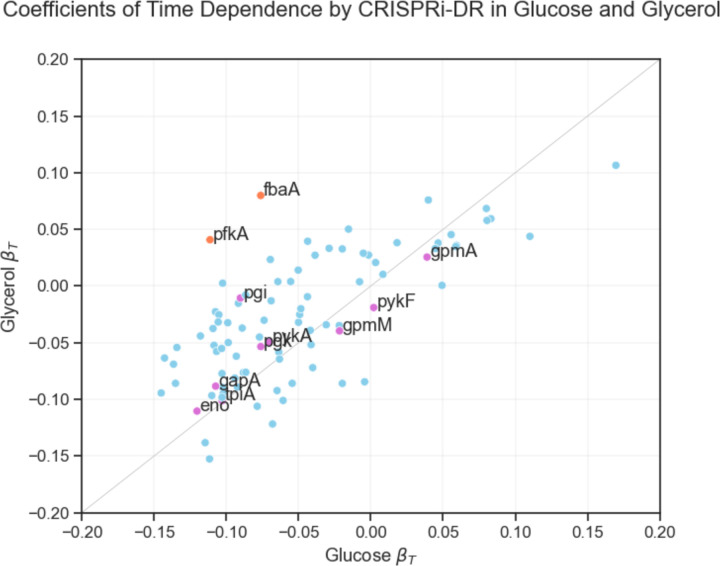
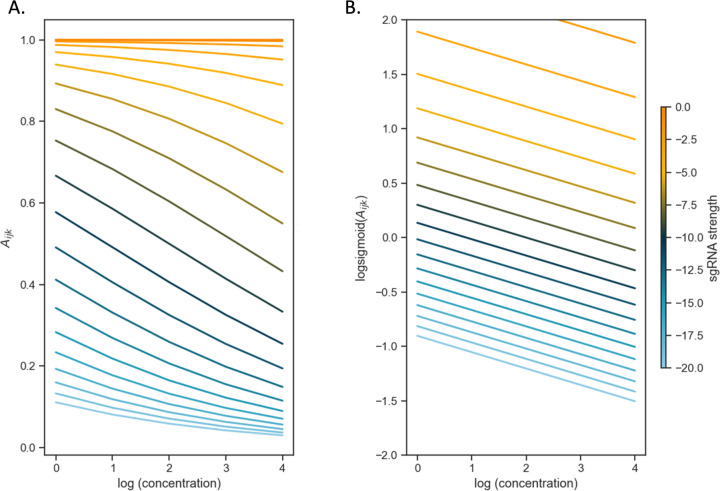
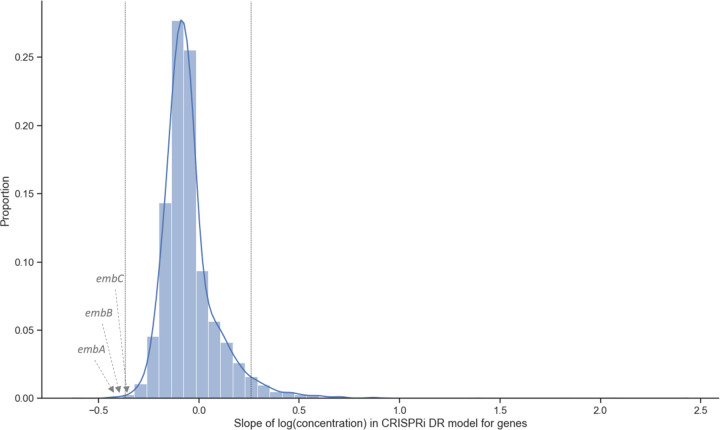
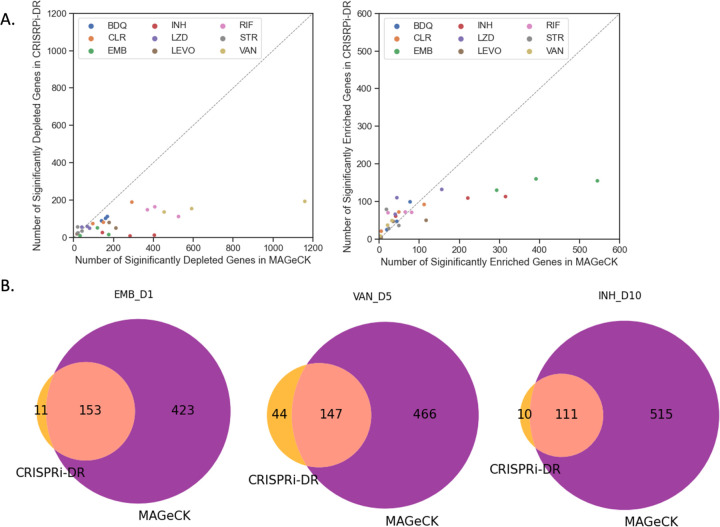
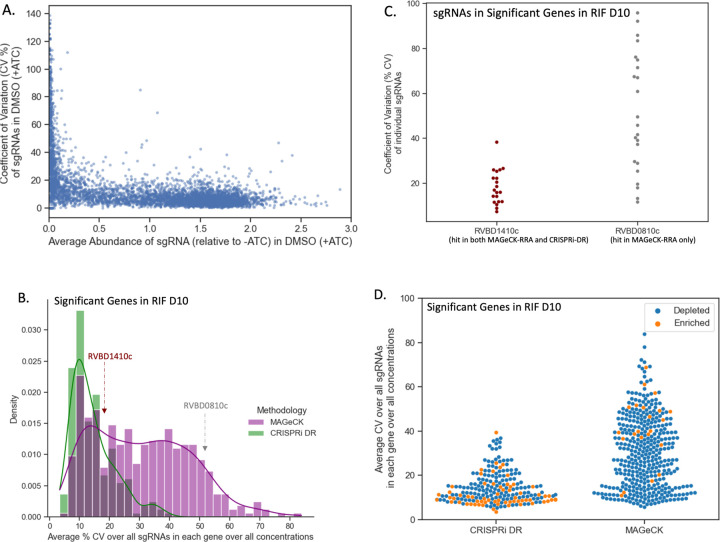
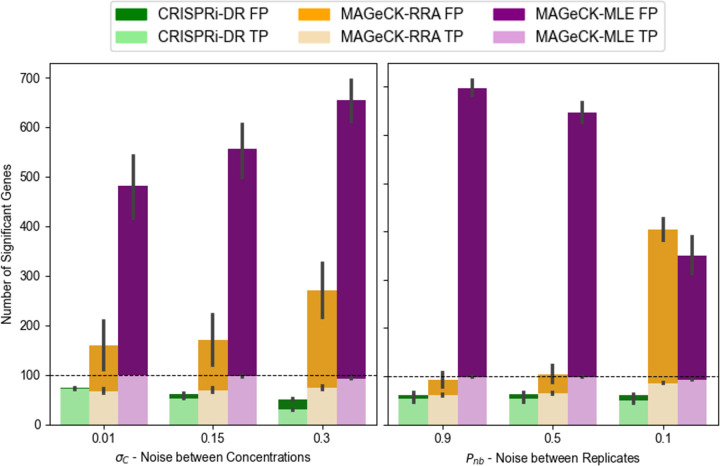
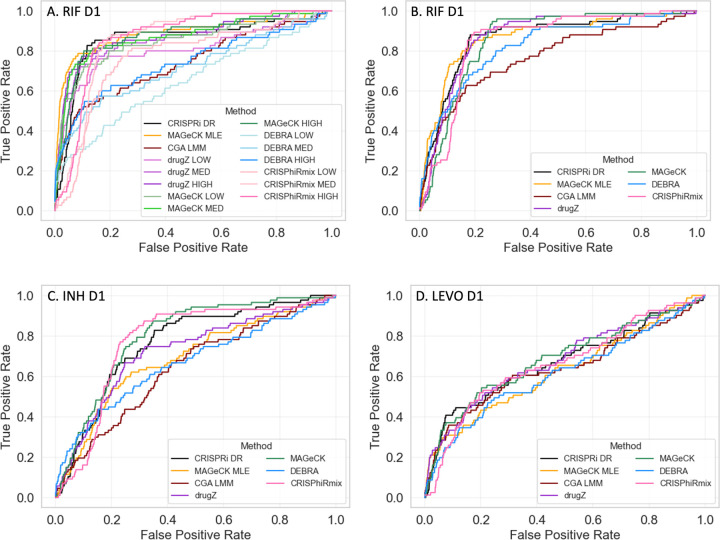
An important application of CRISPR interference (CRISPRi) technology is for identifying chemical-genetic interactions (CGIs). Discovery of genes that interact with exposure to antibiotics can yield insights to drug targets and mechanisms of action or resistance. The objective is to identify CRISPRi mutants whose relative abundance is suppressed (or enriched) in the presence of a drug when the target protein is depleted, reflecting synergistic behavior. Different sgRNAs for a given target can induce a wide range of protein depletion and differential effects on growth rate. The effect of sgRNA strength can be partially predicted based on sequence features. However, the actual growth phenotype depends on the sensitivity of cells to depletion of the target protein. For essential genes, sgRNA efficiency can be empirically measured by quantifying effects on growth rate. We observe that the most efficient sgRNAs are not always optimal for detecting synergies with drugs. sgRNA efficiency interacts in a non-linear way with drug sensitivity, producing an effect where the concentration-dependence is maximized for sgRNAs of intermediate strength (and less so for sgRNAs that induce too much or too little target depletion). To capture this interaction, we propose a novel statistical method called CRISPRi-DR (for Dose-Response model) that incorporates both sgRNA efficiencies and drug concentrations in a modified dose-response equation. We use CRISPRi-DR to re-analyze data from a recent CGI experiment in to identify genes that interact with antibiotics. This approach can be generalized to non-CGI datasets, which we show via an CRISPRi dataset for growth on different carbon sources. The performance is competitive with the best of several related analytical methods. However, for noisier datasets, some of these methods generate far more significant interactions, likely including many false positives, whereas CRISPRi-DR maintains higher precision, which we observed in both empirical and simulated data.
CRISPR干扰(CRISPRi)技术的一个重要应用是识别化学-遗传相互作用(CGI)。发现与接触抗生素相互作用的基因可以深入了解药物靶点、作用机制或耐药性。目标是识别在靶蛋白缺失时,其相对丰度在药物存在下被抑制(或富集)的CRISPRi突变体,这反映了协同行为。针对给定靶点的不同sgRNA可以诱导广泛的蛋白质缺失以及对生长速率的不同影响。sgRNA强度的影响可以部分基于序列特征进行预测。然而,实际的生长表型取决于细胞对靶蛋白缺失的敏感性。对于必需基因,sgRNA效率可以通过量化对生长速率的影响来进行经验性测量。我们观察到,最有效的sgRNA并不总是检测与药物协同作用的最佳选择。sgRNA效率与药物敏感性以非线性方式相互作用,产生一种效应,即中等强度的sgRNA浓度依赖性最大(而诱导过多或过少靶蛋白缺失的sgRNA浓度依赖性较小)。为了捕捉这种相互作用,我们提出了一种名为CRISPRi-DR(剂量-反应模型)的新型统计方法,该方法在修正的剂量-反应方程中纳入了sgRNA效率和药物浓度。我们使用CRISPRi-DR重新分析了最近一项关于识别与抗生素相互作用基因的CGI实验的数据。这种方法可以推广到非CGI数据集,我们通过一个关于在不同碳源上生长的CRISPRi数据集展示了这一点。其性能与几种相关分析方法中的最佳方法具有竞争力。然而,对于噪声更大的数据集,其中一些方法会产生更多显著的相互作用,可能包括许多假阳性,而CRISPRi-DR保持了更高的精度,这在经验数据和模拟数据中我们都观察到了。