Molecular Genetics and Genetic Diagnostic Units, Institute of Rare Diseases Research (IIER), Spanish National Institute of Health Carlos III (ISCIII), 28220 Madrid, Spain.
CIBER de Enfermedades Raras, CIBERER U758, 28029 Madrid, Spain.
Int J Mol Sci. 2023 Aug 10;24(16):12645. doi: 10.3390/ijms241612645.
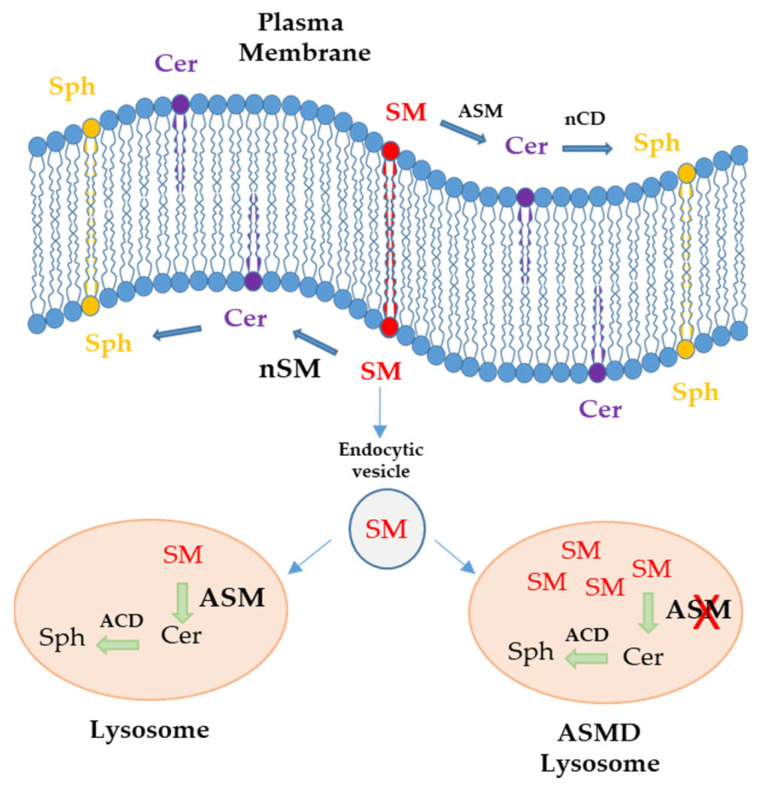
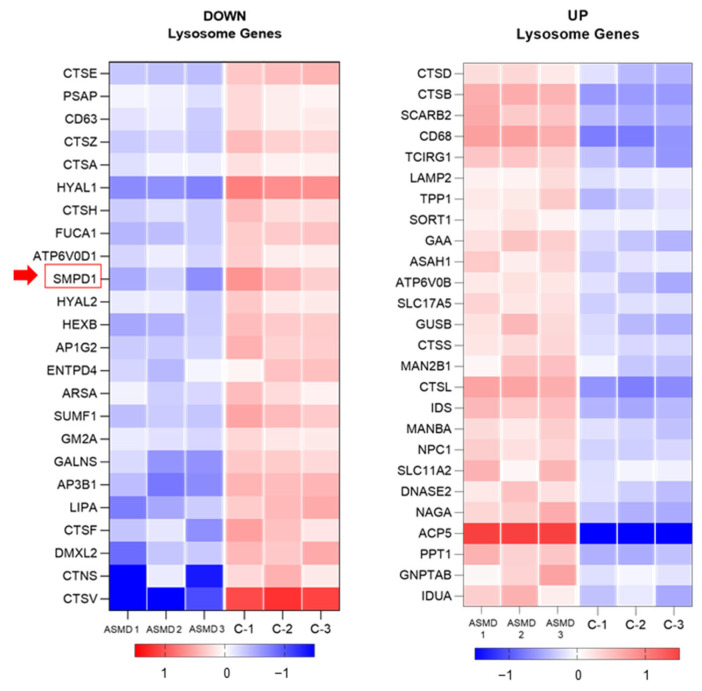
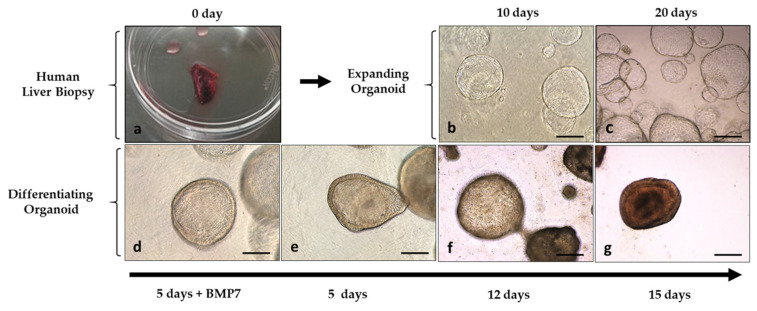
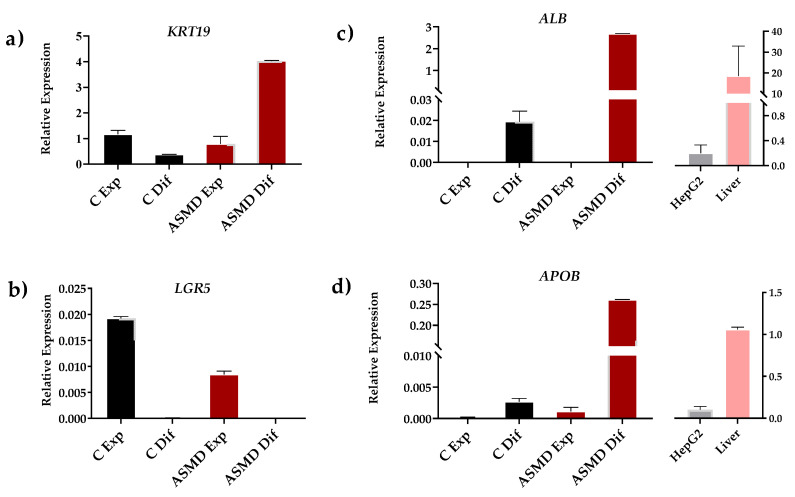
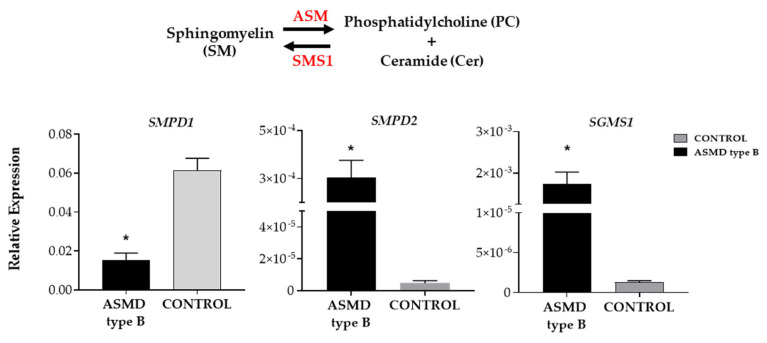
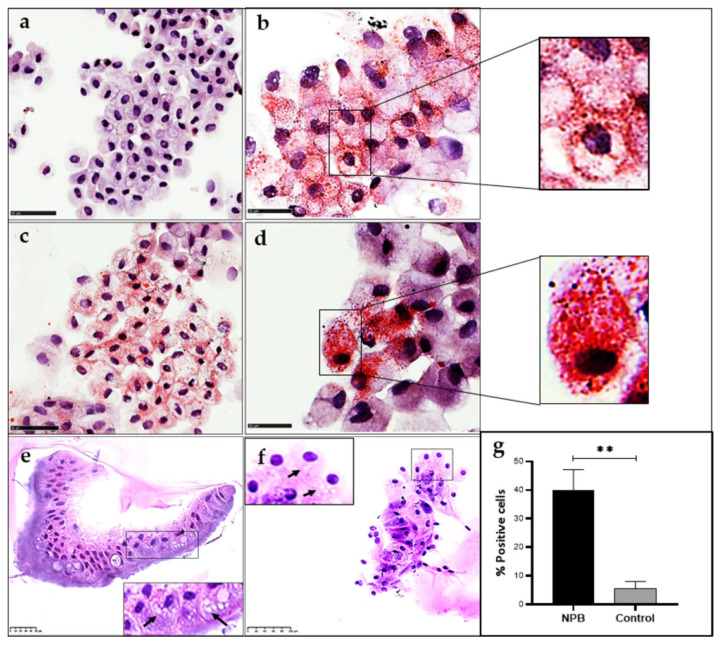
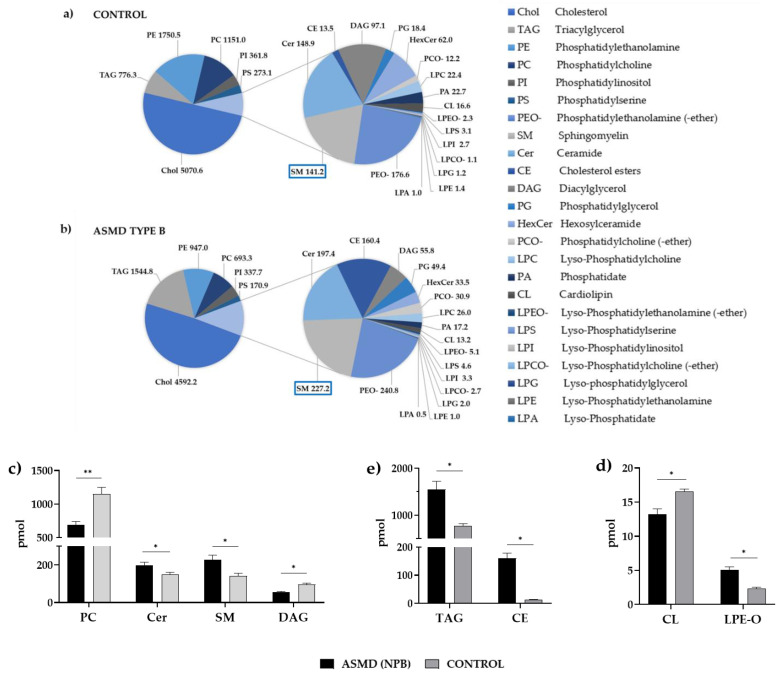
Acid sphingomyelinase deficiency (ASMD) or Niemann-Pick disease type A (NPA), type B (NPB) and type A/B (NPA/B), is a rare lysosomal storage disease characterized by progressive accumulation of sphingomyelin (SM) in the liver, lungs, bone marrow and, in severe cases, neurons. A disease model was established by generating liver organoids from a NPB patient carrying the p.Arg610del variant in the gene. Liver organoids were characterized by transcriptomic and lipidomic analysis. We observed altered lipid homeostasis in the patient-derived organoids showing the predictable increase in sphingomyelin (SM), together with cholesterol esters (CE) and triacylglycerides (TAG), and a reduction in phosphatidylcholine (PC) and cardiolipins (CL). Analysis of lysosomal gene expression pointed to 24 downregulated genes, including , and 26 upregulated genes that reflect the lysosomal stress typical of the disease. Altered genes revealed reduced expression of enzymes that could be involved in the accumulation in the hepatocytes of sphyngoglycolipids and glycoproteins, as well as upregulated genes coding for different glycosidases and cathepsins. Lipidic and transcriptome changes support the use of hepatic organoids as ideal models for ASMD investigation.
酸性鞘磷脂酶缺乏症(ASMD)或尼曼-匹克病 A 型(NPA)、B 型(NPB)和 A/B 型(NPA/B),是一种罕见的溶酶体贮积病,其特征是鞘磷脂(SM)在肝脏、肺部、骨髓中进行性积累,在严重的情况下,还会在神经元中积累。通过从携带基因 p.Arg610del 变异的 NPB 患者中生成肝类器官,建立了一种疾病模型。通过转录组学和脂质组学分析对肝类器官进行了特征描述。我们观察到,在源自患者的类器官中,脂质稳态发生了改变,表现为鞘磷脂(SM)、胆固醇酯(CE)和三酰基甘油(TAG)的可预测增加,同时磷酸胆碱(PC)和心磷脂(CL)减少。溶酶体基因表达分析表明,有 24 个下调基因,包括 、和 ,以及 26 个上调基因,反映了疾病特有的溶酶体应激。改变的基因显示出参与鞘糖脂和糖蛋白在肝细胞中积累的酶的表达减少,以及编码不同糖苷酶和组织蛋白酶的上调基因。脂质和转录组变化支持将肝类器官用作 ASMD 研究的理想模型。