Department of Physiology, Faculty of Medicine, University of Debrecen, Debrecen, Hungary.
Department of Orthodontics, Faculty of Dentistry, University of Debrecen, Debrecen, Hungary.
Sci Rep. 2023 Sep 5;13(1):14659. doi: 10.1038/s41598-023-41801-2.
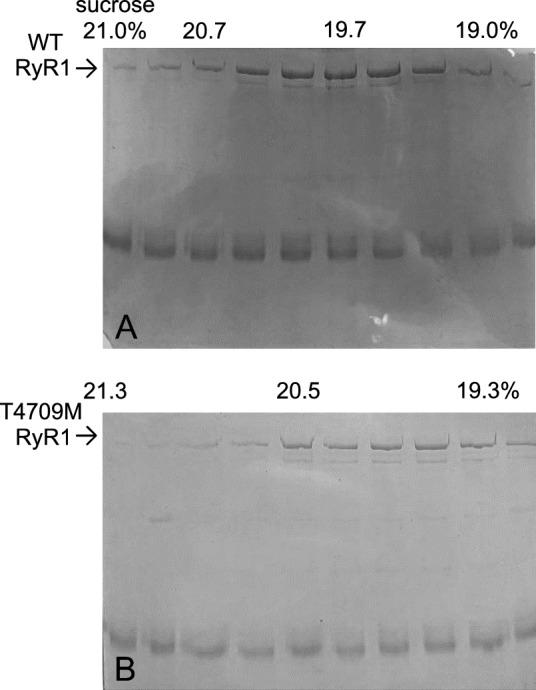
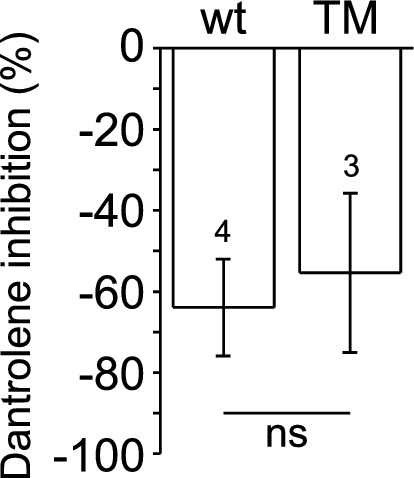
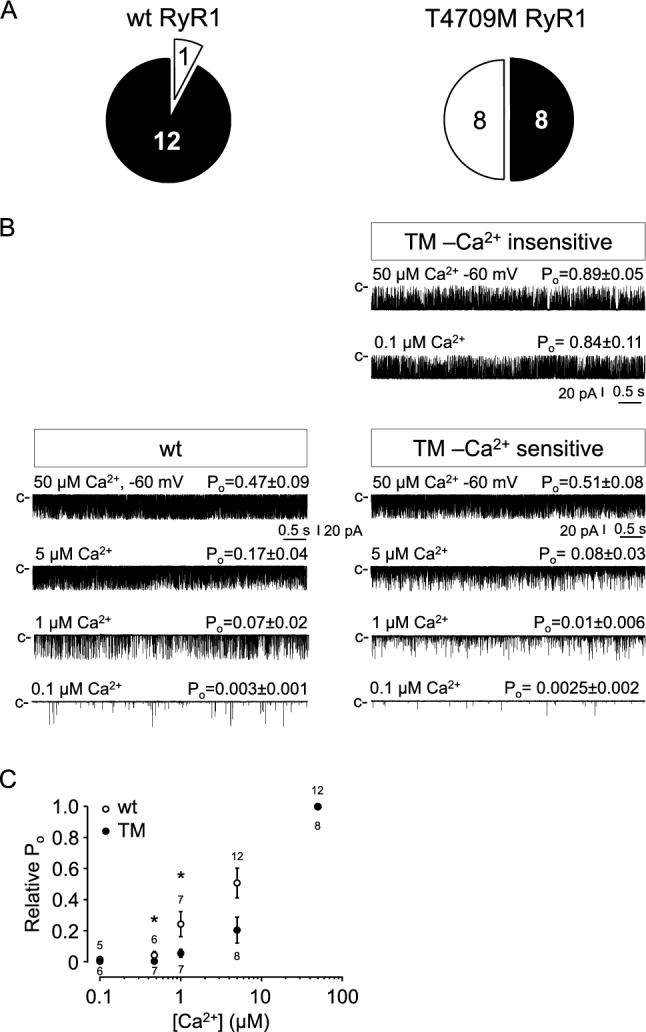
Physiological muscle contraction requires an intact ligand gating mechanism of the ryanodine receptor 1 (RyR1), the Ca-release channel of the sarcoplasmic reticulum. Some mutations impair the gating and thus cause muscle disease. The RyR1 mutation T4706M is linked to a myopathy characterized by muscle weakness. Although, low expression of the T4706M RyR1 protein can explain in part the symptoms, little is known about the function RyR1 channels with this mutation. In order to learn whether this mutation alters channel function in a manner that can account for the observed symptoms, we examined RyR1 channels isolated from mice homozygous for the T4709M (TM) mutation at the single channel level. Ligands, including Ca, ATP, Mg and the RyR inhibitor dantrolene were tested. The full conductance of the TM channel was the same as that of wild type (wt) channels and a population of partial open (subconductive) states were not observed. However, two unique sub-populations of TM RyRs were identified. One half of the TM channels exhibited high open probability at low (100 nM) and high (50 μM) cytoplasmic [Ca], resulting in Ca-insensitive, constitutively high P channels. The rest of the TM channels exhibited significantly lower activity within the physiologically relevant range of cytoplasmic [Ca], compared to wt. TM channels retained normal Mg block, modulation by ATP, and inhibition by dantrolene. Together, these results suggest that the TM mutation results in a combination of primary and secondary RyR1 dysfunctions that contribute to disease pathogenesis.
生理肌肉收缩需要完整的兰尼碱受体 1(RyR1)配体门控机制,RyR1 是肌质网的 Ca2+释放通道。某些突变会损害门控作用,从而导致肌肉疾病。RyR1 突变 T4706M 与一种以肌肉无力为特征的肌病有关。尽管 T4706M RyR1 蛋白的低表达部分可以解释这些症状,但人们对这种突变的 RyR1 通道的功能知之甚少。为了了解该突变是否以可以解释观察到的症状的方式改变通道功能,我们在单个通道水平上检查了 T4709M(TM)突变纯合的小鼠中的 RyR1 通道。测试了配体,包括 Ca2+、ATP、Mg2+和 RyR 抑制剂丹曲林。TM 通道的全电导与野生型(wt)通道相同,并且未观察到部分开放(亚电导)状态的群体。然而,鉴定出 TM RyR 的两个独特的亚群。TM 通道的一半在低(100 nM)和高(50 μM)细胞质 [Ca2+] 下表现出高的开放概率,导致 Ca2+不敏感的、组成性的高 P 通道。TM 通道的其余部分在与生理相关的细胞质 [Ca2+] 范围内表现出明显较低的活性,与 wt 相比。TM 通道保留了正常的 Mg2+阻断、ATP 调节和丹曲林抑制。总之,这些结果表明 TM 突变导致原发性和继发性 RyR1 功能障碍的组合,这有助于疾病的发病机制。