Wang Yao, Pang Bo, Wang Zequn, Tian Xueying, Xu Xiaoying, Chong Xiaowen, Liang Hao, Ma Wei, Kou Zengqiang, Wen Hongling
Department of Epidemiology, School of Public Health, Cheeloo College of Medicine, Shandong University, Jinan, China.
Department of Microbiological Laboratory Technology, School of Public Health, Cheeloo College of Medicine, Shandong University, Jinan, China.
Front Microbiol. 2023 Aug 21;14:1233693. doi: 10.3389/fmicb.2023.1233693. eCollection 2023.
Conducting an up-to-date analysis on the genomic diversity and evolution patterns of severe fever with thrombocytopenia syndrome virus (SFTSV) is crucial for elucidating the underlying mechanisms of its emergency and pathogenicity, as well as assessing the extent of its threat to public health.
Complete genome sequences of SFTSV were obtained from GenBank until December 19, 2022. A thorough phylogenetic analysis was conducted using comprehensive bioinformatics methods to estimate the genomic diversity and evolution.
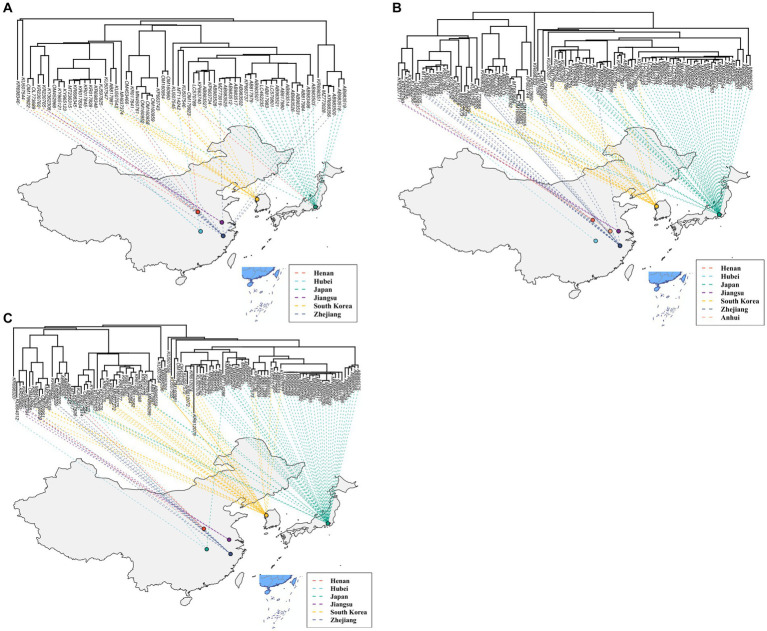
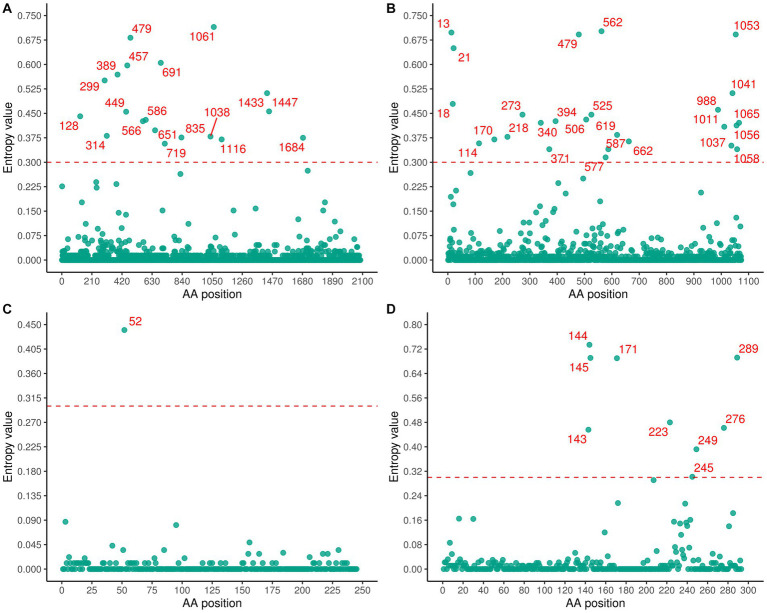
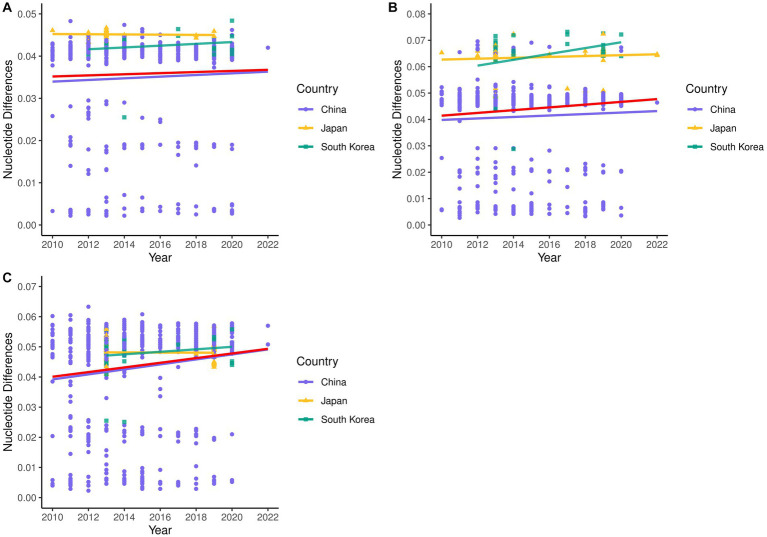
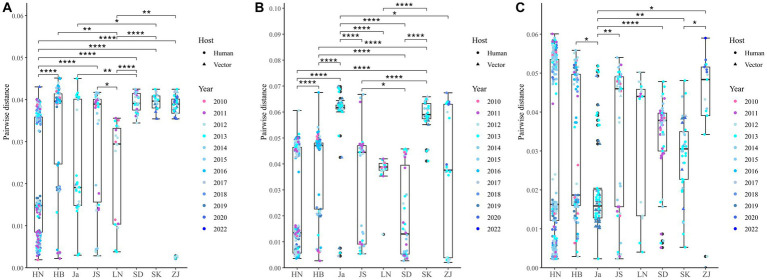
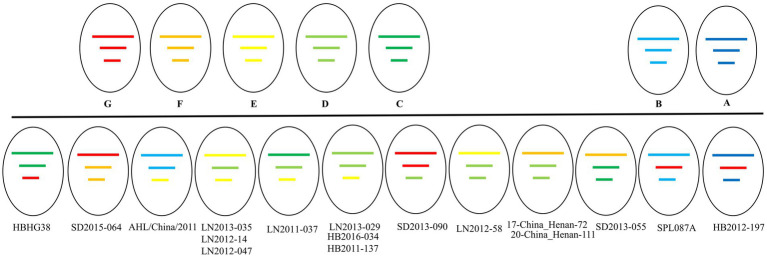
The phylogenetic classification of SFTSV strains yielded seven lineages (A-G) for each genome segment. SFTSV displayed notable variations in evolutionary patterns among different regions and segments, without a linear accumulation of nucleotide substitutions within segments and regions. The comprehensive analysis revealed 54 recombination events and 17 reassortment strains, including the first discovery of recombination events involving sea-crossing and species-crossing. Selection analysis identified three positive sites (2, 671, 1353) in RNA-dependent RNA polymerase, three positive sites (22, 298, 404) in glycoprotein, and two positive sites (9, 289) in nonstructural protein. No positive selection sites were found in nucleoprotein.
Our study unveiled the existence of multiple evolutionary forces influencing SFTSV, contributing to its increasing genetic diversity, which had the potential to modify its antigenicity and pathogenicity. Furthermore, our study highlights the importance of tracking the spread of SFTSV across regions and species.
对发热伴血小板减少综合征病毒(SFTSV)的基因组多样性和进化模式进行最新分析,对于阐明其暴发及致病性的潜在机制,以及评估其对公共卫生的威胁程度至关重要。
从GenBank获取截至2022年12月19日的SFTSV完整基因组序列。使用综合生物信息学方法进行全面的系统发育分析,以估计基因组多样性和进化情况。
SFTSV毒株的系统发育分类在每个基因组片段中产生了7个谱系(A - G)。SFTSV在不同区域和片段之间的进化模式存在显著差异,片段和区域内没有核苷酸替换的线性积累。综合分析揭示了54个重组事件和17个重配毒株,包括首次发现涉及跨海域和跨物种的重组事件。选择分析在RNA依赖的RNA聚合酶中鉴定出3个阳性位点(2、671、1353),在糖蛋白中鉴定出3个阳性位点(22、298、404),在非结构蛋白中鉴定出2个阳性位点(9、289)。在核蛋白中未发现阳性选择位点。
我们的研究揭示了影响SFTSV的多种进化力量的存在,这导致其遗传多样性增加,有可能改变其抗原性和致病性。此外,我们的研究强调了追踪SFTSV在区域和物种间传播的重要性。