Department of Chemistry, University of Wisconsin, 1101 University Avenue, Madison, Wisconsin 53706, United States.
Department of Chemistry, The Scripps Research Institute, La Jolla, California 92037, United States.
J Am Chem Soc. 2023 Sep 20;145(37):20539-20550. doi: 10.1021/jacs.3c06703. Epub 2023 Sep 11.
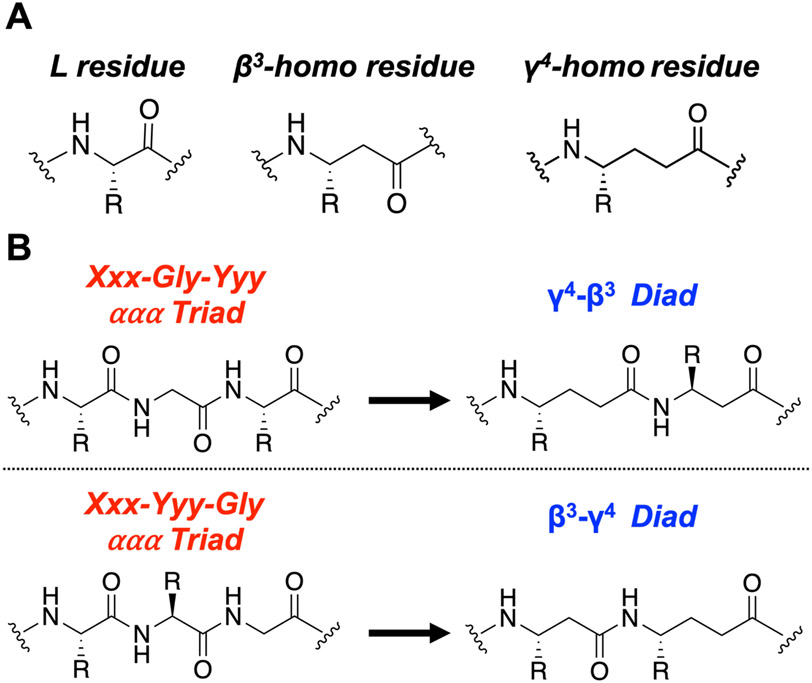
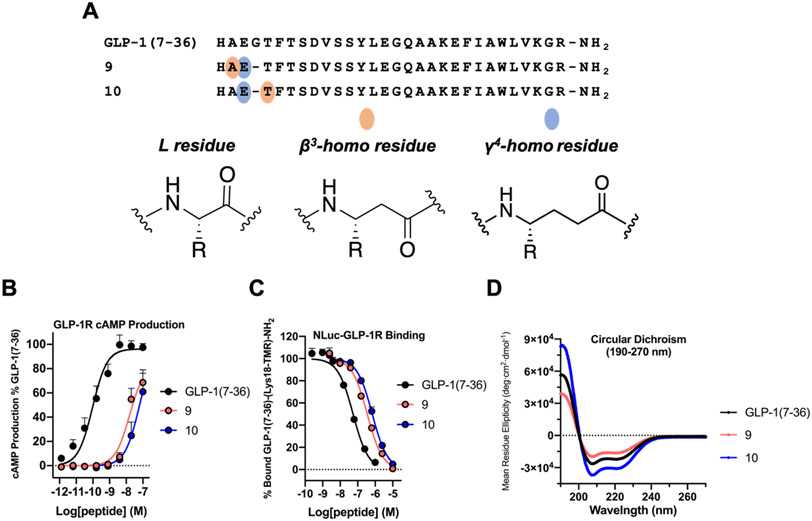
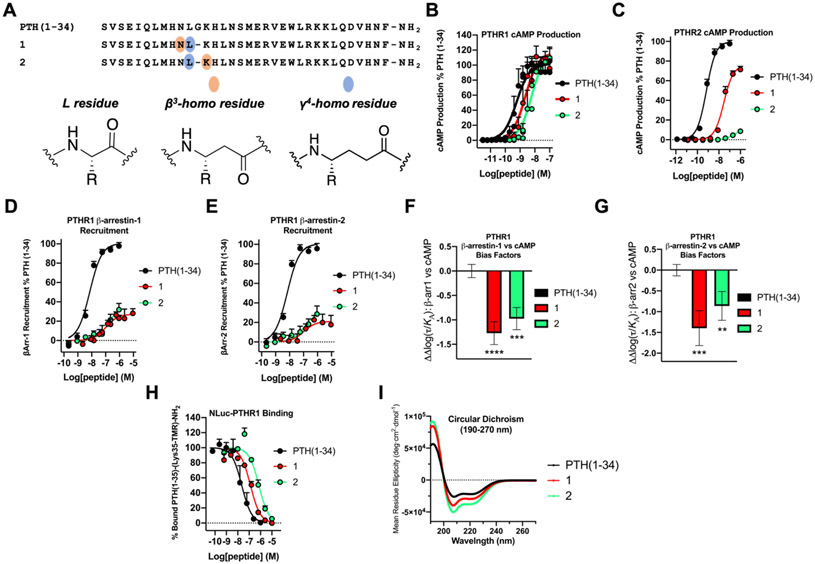
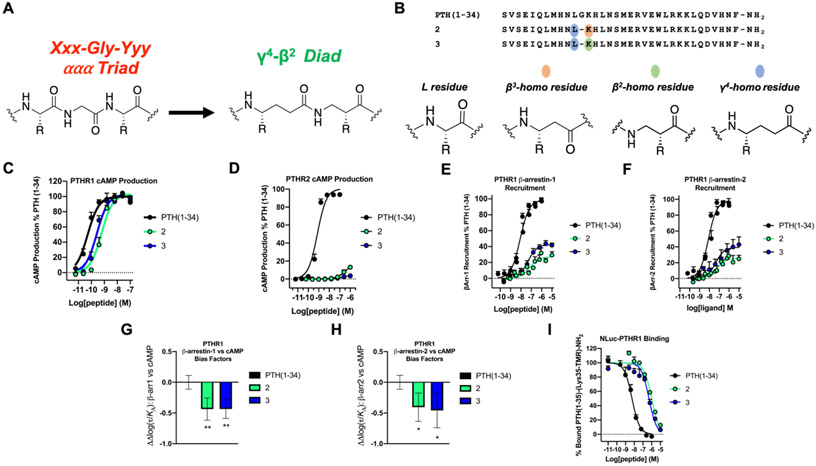
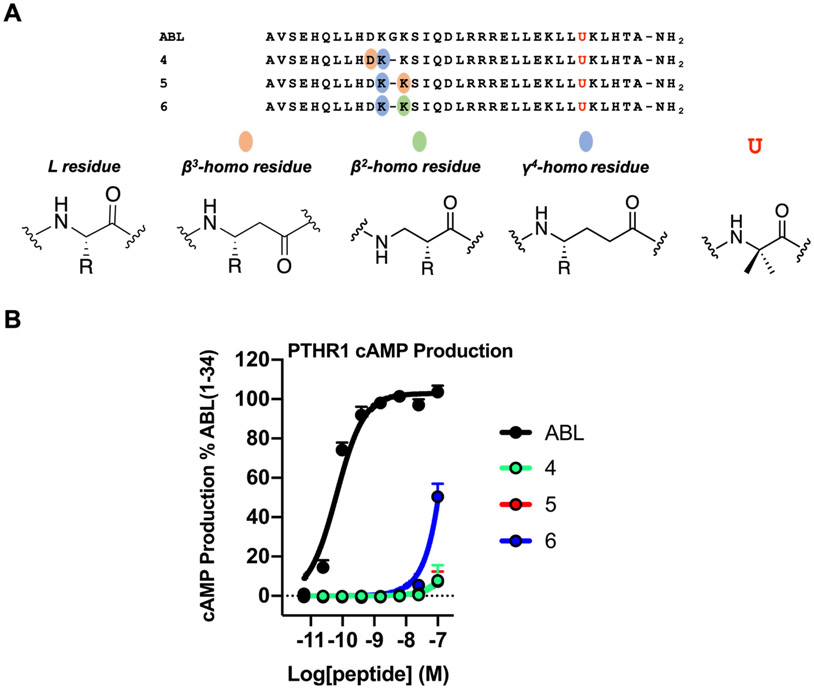
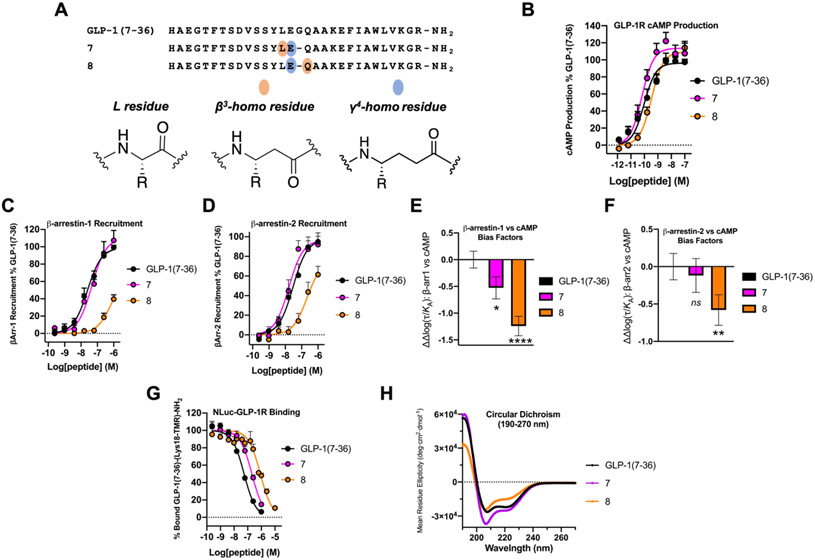
We have applied an underexplored backbone modification strategy to generate new analogues of peptides that activate two clinically important class B1 G protein-coupled receptors (GPCRs). Most peptide modification strategies involve changing side chains or, less commonly, changing the configuration at side chain-bearing carbons (i.e., l residues replaced by d residues). In contrast, backbone modifications alter the number of backbone atoms and the identities of backbone atoms relative to a poly-α-amino acid backbone. Starting from the peptide agonists PTH(1-34) (the first 34 residues of the parathyroid hormone, used clinically as the drug teriparatide) and glucagon-like peptide-1 (7-36) (GLP-1(7-36)), we replaced native α-residue triads with a diad composed of a β-amino acid residue and a γ-amino acid residue. The β/γ diad retains the number of backbone atoms in the ααα triad. Because the β and γ residue each bear a single side chain, we implemented ααα→βγ replacements at sites that contained a Gly residue (i.e., at α-residue triads that presented only two side chains). All seven of the α/β/γ-peptides derived from PTH(1-34) or GLP-1(7-36) bind to the cognate receptor (the PTHR1 or the GLP-1R), but they vary considerably in their activity profiles. Outcomes include functional mimicry of the all-α agonist, receptor-selective agonist activity, biased agonism, or strong binding with weak activation, which could lead to antagonist development. Collectively, these findings demonstrate that ααα→βγ replacements, which are easily implemented via solid-phase synthesis, can generate peptide hormone analogues that display unique and potentially useful signaling behavior.
我们应用了一种尚未充分探索的骨架修饰策略,来生成能激活两种临床上重要的 B1 类 G 蛋白偶联受体(GPCR)的新型肽类似物。大多数肽修饰策略涉及改变侧链,或者较少见地,改变带有侧链的碳原子的构型(即,l 残基被 d 残基取代)。相比之下,骨架修饰会改变骨架原子的数量和相对于聚-α-氨基酸骨架的骨架原子的身份。从肽激动剂 PTH(1-34)(甲状旁腺激素的前 34 个残基,临床上用作药物特立帕肽)和胰高血糖素样肽-1(7-36)(GLP-1(7-36))开始,我们用由β-氨基酸残基和γ-氨基酸残基组成的二联体取代了天然的α-残基三联体。β/γ二联体保留了ααα三联体中的骨架原子数量。由于β和γ残基各带有一个侧链,因此我们在含有甘氨酸残基的位点(即,仅带有两条侧链的α-残基三联体)实施了ααα→βγ替换。从 PTH(1-34)或 GLP-1(7-36)衍生的 7 种α/β/γ-肽都与相应的受体(PTHR1 或 GLP-1R)结合,但它们的活性谱差异很大。结果包括对全-α激动剂的功能模拟、受体选择性激动剂活性、偏激动剂作用或强结合弱激活,这可能导致拮抗剂的开发。总的来说,这些发现表明,通过固相合成很容易实现的ααα→βγ替换可以生成具有独特且可能有用的信号转导行为的肽激素类似物。