Bohn Emma, Lau Tammy T Y, Wagih Omar, Masud Tehmina, Merico Daniele
Deep Genomics Inc., Toronto, ON, Canada.
The Centre for Applied Genomics, Hospital for Sick Children, Toronto, ON, Canada.
Front Mol Biosci. 2023 Sep 8;10:1257550. doi: 10.3389/fmolb.2023.1257550. eCollection 2023.
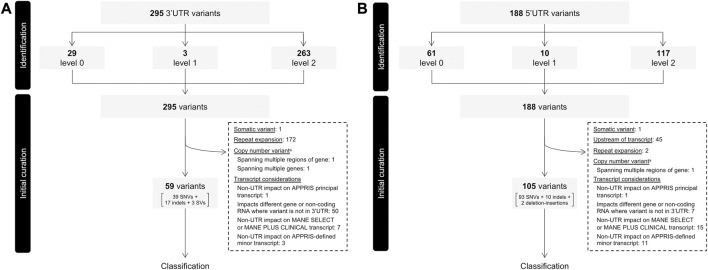
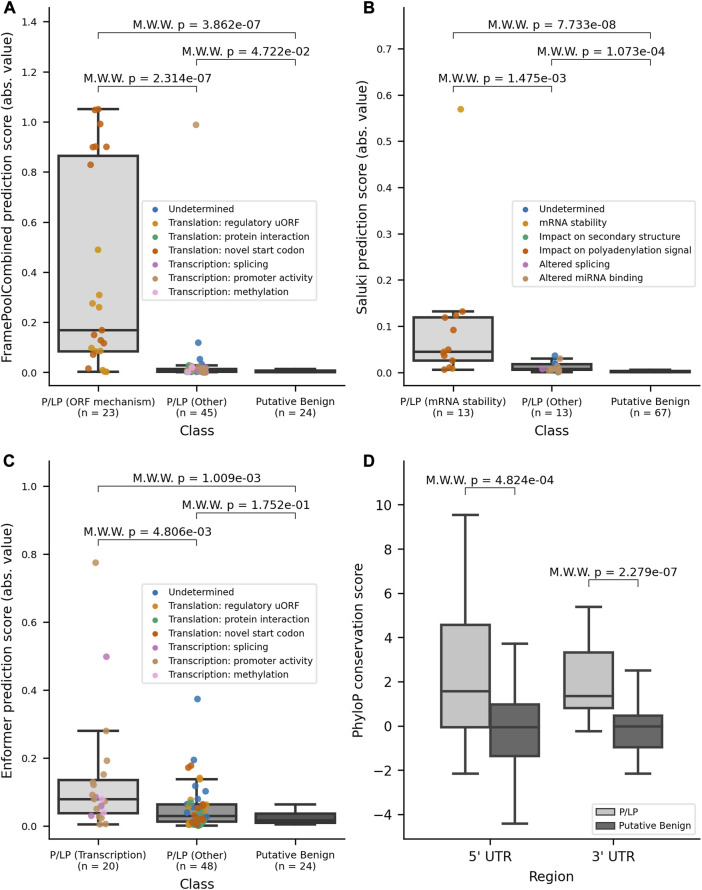
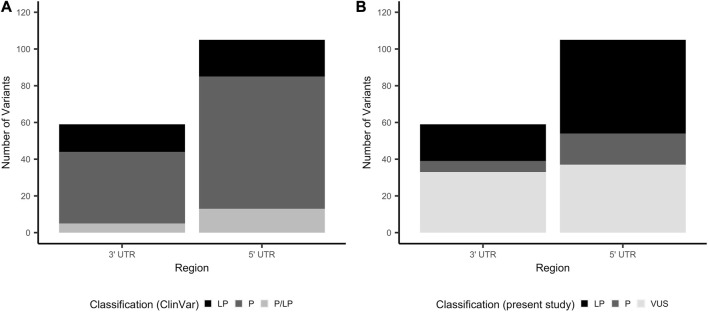
Variants in 5' and 3' untranslated regions (UTR) contribute to rare disease. While predictive algorithms to assist in classifying pathogenicity can potentially be highly valuable, the utility of these tools is often unclear, as it depends on carefully selected training and validation conditions. To address this, we developed a high confidence set of pathogenic (P) and likely pathogenic (LP) variants and assessed deep learning (DL) models for predicting their molecular effects. 3' and 5' UTR variants documented as P or LP (P/LP) were obtained from ClinVar and refined by reviewing the annotated variant effect and reassessing evidence of pathogenicity following published guidelines. Prediction scores from sequence-based DL models were compared between three groups: P/LP variants acting though the mechanism for which the model was designed (model-matched), those operating through other mechanisms (model-mismatched), and putative benign variants. PhyloP was used to compare conservation scores between P/LP and putative benign variants. 295 3' and 188 5' UTR variants were obtained from ClinVar, of which 26 3' and 68 5' UTR variants were classified as P/LP. Predictions by DL models achieved statistically significant differences when comparing modelmatched P/LP variants to both putative benign variants and modelmismatched P/LP variants, as well as when comparing all P/LP variants to putative benign variants. PhyloP conservation scores were significantly higher among P/LP compared to putative benign variants for both the 3' and 5' UTR. In conclusion, we present a high-confidence set of P/LP 3' and 5' UTR variants spanning a range of mechanisms and supported by detailed pathogenicity and molecular mechanism evidence curation. Predictions from DL models further substantiate these classifications. These datasets will support further development and validation of DL algorithms designed to predict the functional impact of variants that may be implicated in rare disease.
5'和3'非翻译区(UTR)的变异会导致罕见病。虽然有助于致病性分类的预测算法可能具有很高的价值,但这些工具的实用性往往不明确,因为这取决于精心选择的训练和验证条件。为了解决这个问题,我们开发了一组高置信度的致病性(P)和可能致病性(LP)变异,并评估了用于预测其分子效应的深度学习(DL)模型。从ClinVar获得记录为P或LP(P/LP)的3'和5'UTR变异,并通过审查注释的变异效应和按照已发表的指南重新评估致病性证据进行完善。在三组之间比较基于序列的DL模型的预测分数:通过模型设计机制起作用的P/LP变异(模型匹配)、通过其他机制起作用的变异(模型不匹配)和推定的良性变异。使用PhyloP比较P/LP和推定的良性变异之间的保守分数。从ClinVar获得了295个3'UTR变异和188个5'UTR变异,其中26个3'UTR变异和68个5'UTR变异被分类为P/LP。当将模型匹配的P/LP变异与推定的良性变异和模型不匹配的P/LP变异进行比较时,以及将所有P/LP变异与推定的良性变异进行比较时,DL模型的预测实现了统计学上的显著差异。对于3'和5'UTR,P/LP中的PhyloP保守分数显著高于推定的良性变异。总之,我们展示了一组高置信度的3'和5'UTR P/LP变异,涵盖一系列机制,并得到详细的致病性和分子机制证据整理的支持。DL模型的预测进一步证实了这些分类。这些数据集将支持旨在预测可能与罕见病相关的变异功能影响的DL算法的进一步开发和验证。