CIBERER, Center for Biomedical Research in Rare Diseases Network, 28029 Madrid, Spain.
INGEMM-IdiPaz, Institute of Medical and Molecular Genetics, 28046 Madrid, Spain.
Genes (Basel). 2023 Aug 23;14(9):1664. doi: 10.3390/genes14091664.
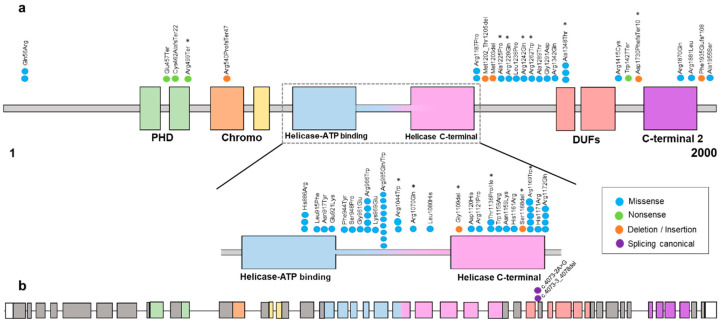
Snijders Blok-Campeau syndrome (SNIBCPS, OMIM# 618205) is an extremely infrequent disease with only approximately 60 cases reported so far. SNIBCPS belongs to the group of neurodevelopmental disorders (NDDs). Clinical features of patients with SNIBCPS include global developmental delay, intellectual disability, speech and language difficulties and behavioral disorders like autism spectrum disorder. In addition, patients with SNIBCPS exhibit typical dysmorphic features including macrocephaly, hypertelorism, sparse eyebrows, broad forehead, prominent nose and pointed chin. The severity of the neurological effects as well as the presence of other features is variable among subjects. SNIBCPS is caused likely by pathogenic and pathogenic variants in , which seems to be involved in chromatin remodeling by deacetylating histones. Here, we report 20 additional patients with clinical features compatible with SNIBCPS from 17 unrelated families with confirmed likely pathogenic/pathogenic variants in . Patients were analyzed by whole exome sequencing and segregation studies were performed by Sanger sequencing. Patients in this study showed different pathogenic variants affecting several functional domains of the protein. Additionally, none of the variants described here were reported in control population databases, and most computational predictors suggest that they are deleterious. The most common clinical features of the whole cohort of patients are global developmental delay (98%) and speech disorder/delay (92%). Other frequent features (51-74%) include intellectual disability, hypotonia, hypertelorism, abnormality of vision, macrocephaly and prominent forehead, among others. This study expands the number of individuals with confirmed SNIBCPS due to pathogenic or likely pathogenic variants in Furthermore, we add evidence of the importance of the application of massive parallel sequencing for NDD patients for whom the clinical diagnosis might be challenging and where deep phenotyping is extremely useful to accurately manage and follow up the patients.
斯尼德布洛克-坎波综合征(SNIBCPS,OMIM#618205)是一种极其罕见的疾病,迄今为止,仅有约 60 例报道。SNIBCPS 属于神经发育障碍(NDD)组。SNIBCPS 患者的临床特征包括全面发育迟缓、智力障碍、言语和语言困难以及自闭症谱系障碍等行为障碍。此外,SNIBCPS 患者还表现出典型的发育不良特征,包括大头畸形、远视、稀疏的眉毛、宽阔的额头、突出的鼻子和尖下巴。神经效应的严重程度以及其他特征的存在在受试者之间是不同的。SNIBCPS 可能由 中的致病性和可能致病性变体引起,该基因似乎通过去乙酰化组蛋白参与染色质重塑。在这里,我们报告了来自 17 个无关家族的 20 名具有与 SNIBCPS 相符的临床特征的患者,这些家族均携带 中经证实的可能致病性/致病性变体。通过全外显子组测序对患者进行分析,并通过 Sanger 测序进行遗传分析。本研究中的患者表现出影响蛋白几个功能域的不同致病性变异。此外,这里描述的变异均未在对照人群数据库中报道,并且大多数计算预测器表明它们是有害的。整个患者队列的最常见临床特征是全面发育迟缓(98%)和言语障碍/延迟(92%)。其他常见特征(51%-74%)包括智力障碍、低张力、远视、视力异常、大头畸形和突出的额头等。本研究扩大了由于 中的致病性或可能致病性变体而确诊为 SNIBCPS 的个体数量。此外,我们为神经发育障碍患者应用大规模平行测序提供了证据,对于这些患者,临床诊断可能具有挑战性,并且深度表型分析对于准确管理和随访患者非常有用。