National Institute of Standards and Technology, NIST Charleston, 331 Fort Johnson Road, Charleston, SC 29412, USA.
Genes (Basel). 2023 Aug 25;14(9):1696. doi: 10.3390/genes14091696.
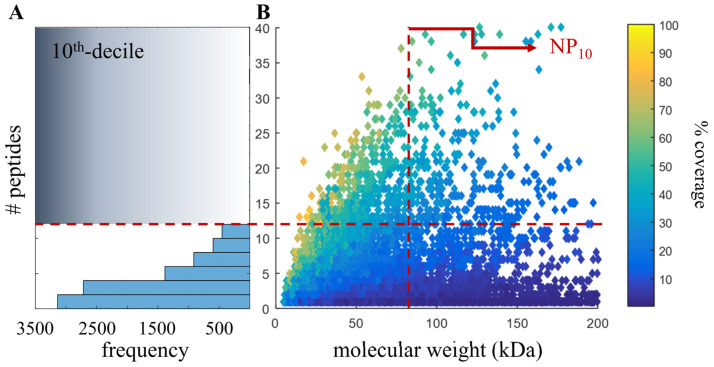
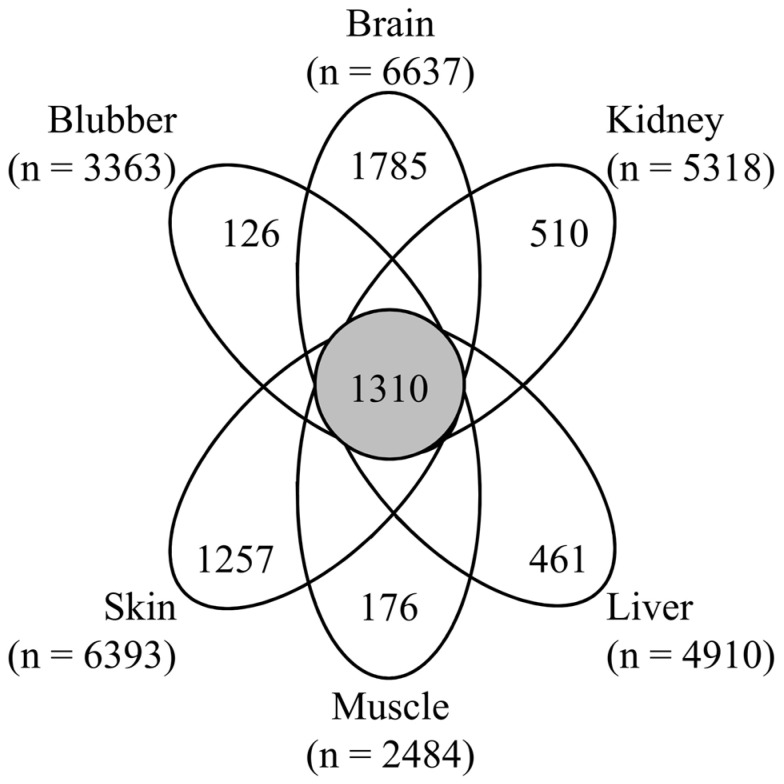
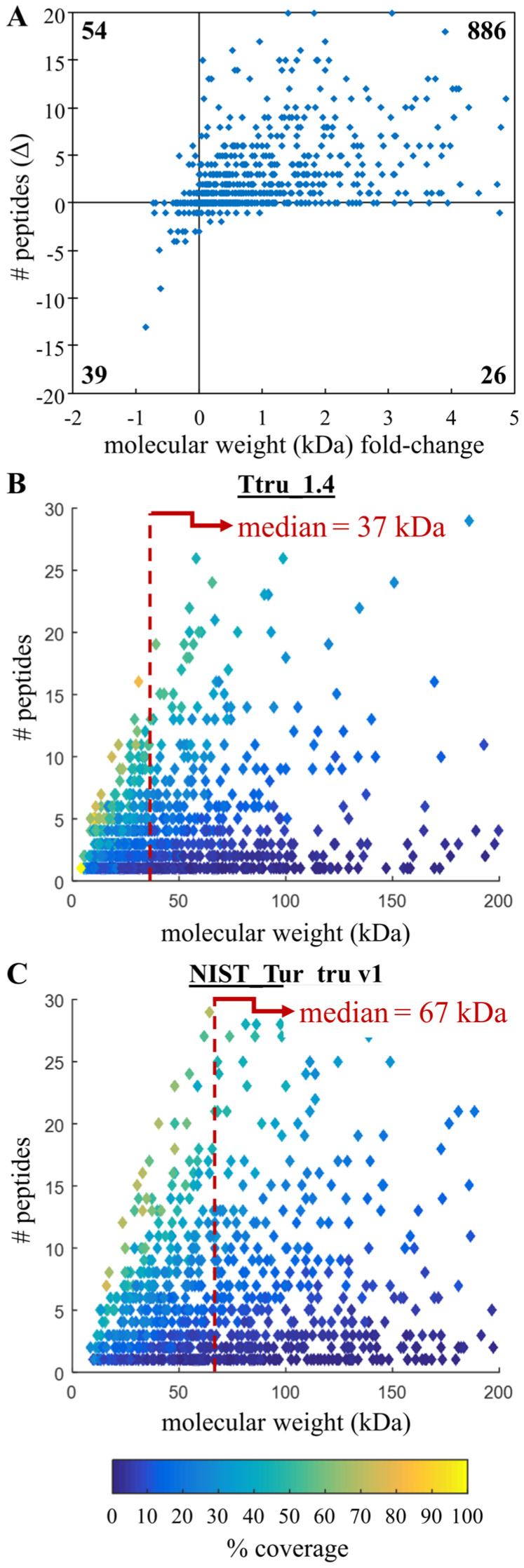
The last decade has witnessed dramatic improvements in whole-genome sequencing capabilities coupled to drastically decreased costs, leading to an inundation of high-quality de novo genomes. For this reason, the continued development of genome quality metrics is imperative. Using the 2016 Atlantic bottlenose dolphin NCBI RefSeq annotation and mass spectrometry-based proteomic analysis of six tissues, we confirmed 10,402 proteins from 4711 protein groups, constituting nearly one-third of the possible predicted proteins. Since the identification of larger proteins with more identified peptides implies reduced database fragmentation and improved gene annotation accuracy, we propose the metric NP, which attempts to capture this quality improvement. The NP metric is calculated by first stratifying proteomic results by identifying the top decile (or 10th 10-quantile) of identified proteins based on the number of peptides per protein and then returns the median molecular weight of the resulting proteins. When using the 2016 versus 2012 genome annotation to search this proteomic data set, there was a 21% improvement in NP. This metric was further demonstrated by using a publicly available proteomic data set to compare human genome annotations from 2004, 2013 and 2016, which showed a 33% improvement in NP. These results demonstrate that proteomics may be a useful metrological tool to benchmark genome accuracy, though there is a need for reference proteomic datasets across species to facilitate the evaluation of new de novo and existing genome.
在过去的十年中,全基因组测序能力得到了显著提高,成本也大幅降低,导致高质量的从头基因组数量激增。因此,继续开发基因组质量指标势在必行。我们使用了 2016 年大西洋宽吻海豚 NCBI RefSeq 注释和基于质谱的六种组织的蛋白质组学分析,从 4711 个蛋白质组中鉴定了 10402 个蛋白质,构成了近三分之一可能预测的蛋白质。由于具有更多鉴定肽的较大蛋白质的鉴定意味着减少了数据库碎片化和提高了基因注释的准确性,因此我们提出了 NP 度量标准,该度量标准试图捕获这种质量改进。NP 度量标准是通过首先根据每个蛋白质的肽数对蛋白质组学结果进行分层,确定鉴定蛋白的前十分位数(或第 10 个十分位数),然后返回所得蛋白质的中值分子量来计算的。当使用 2016 年与 2012 年的基因组注释来搜索这个蛋白质组数据集时,NP 提高了 21%。通过使用公共蛋白质组数据集来比较 2004 年、2013 年和 2016 年的人类基因组注释,进一步证明了这一度量标准,NP 提高了 33%。这些结果表明,蛋白质组学可能是一种有用的计量工具,可以用于基准基因组的准确性,尽管需要跨物种的参考蛋白质组数据集来促进对新的从头和现有的基因组的评估。