Brugnoni Raffaella, Marelli Daria, Iacomino Nicola, Canioni Eleonora, Cappelletti Cristina, Maggi Lorenzo, Ardissone Anna
Neuroimmunology and Neuromuscular Diseases Unit, Department of Clinical Research and Development, Fondazione IRCCS Istituto Neurologico Carlo Besta, 20133 Milan, Italy.
Child Neurology Unit, Department of Pediatric Neuroscience, Fondazione IRCCS Istituto Neurologico Carlo Besta, 20133 Milan, Italy.
Genes (Basel). 2023 Sep 2;14(9):1753. doi: 10.3390/genes14091753.
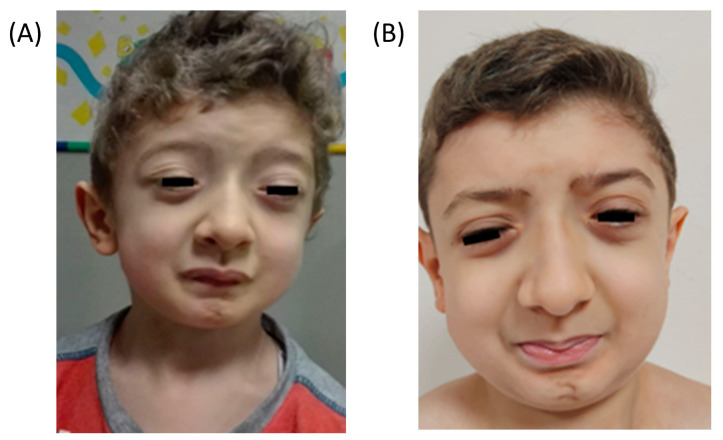
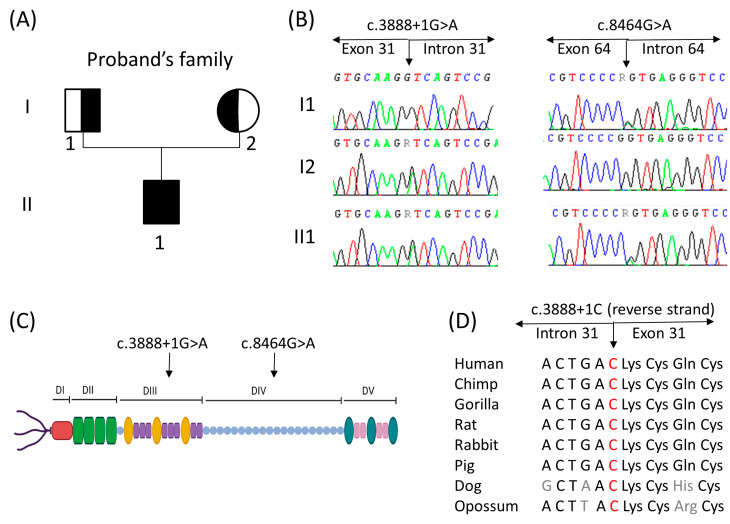
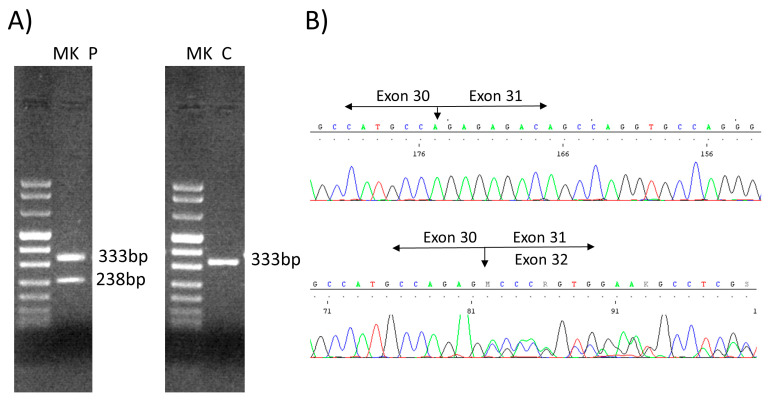
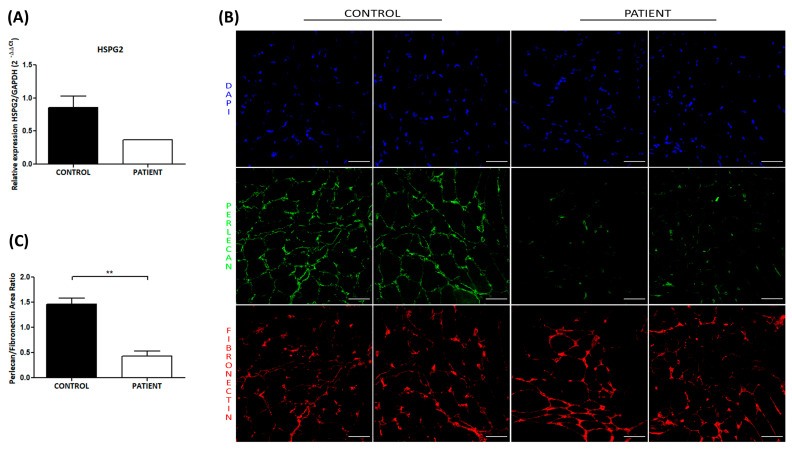
Schwartz-Jampel syndrome type 1 (SJS1) is a rare autosomal recessive musculoskeletal disorder caused by various mutations in the gene encoding the protein perlecan, a major component of basement membranes. We report a novel splice mutation (NM_005529.7):c.3888 + 1G > A and a known point mutation (NM_005529.7):c.8464G > A, leading to the skipping of exon 31 and 64 in mRNA, respectively, in a Moroccan child with clinical features suggestive of SJS1 and carrying two compound heterozygous mutations in the gene detected by next-generation sequencing. Both parents harboured one mutation. Real-time and immunostaining analysis revealed down-regulation of the gene and a mild reduction in the protein in the muscle, respectively. We reviewed all genetically characterized SJS1 cases reported in literature, confirming the clinical hallmarks and unspecific instrumental data in our case. The genotype-phenotype correlation is very challenging in SJS1. Therapy is mainly focused on symptom management and several drugs have been administered with different efficacy.Here, we report the second case with spontaneous improvement.
1型施瓦茨-扬佩尔综合征(SJS1)是一种罕见的常染色体隐性遗传性肌肉骨骼疾病,由编码基底膜主要成分核心蛋白聚糖的基因突变引起。我们报告了一例新的剪接突变(NM_005529.7):c.3888 + 1G > A和一个已知的点突变(NM_005529.7):c.8464G > A,在一名具有SJS1临床特征的摩洛哥儿童中,分别导致mRNA中外显子31和64的跳跃,该儿童通过下一代测序检测到该基因存在两个复合杂合突变。父母双方均携带一个突变。实时分析和免疫染色分析分别显示该基因下调以及肌肉中该蛋白轻度减少。我们回顾了文献中报道的所有经基因鉴定的SJS1病例,证实了我们病例中的临床特征和非特异性检查数据。SJS1中的基因型-表型相关性极具挑战性。治疗主要侧重于症状管理,已使用多种药物,疗效各异。在此,我们报告第二例自发改善的病例。