Department of Microbiology and Immunology and Lineberger Comprehensive Cancer Center, the University of North Carolina at Chapel Hill, Chapel Hill, NC, USA.
Curriculum in Genetics and Molecular Biology, the University of North Carolina at Chapel Hill, Chapel Hill, NC, USA.
Cell Death Dis. 2023 Oct 18;14(10):688. doi: 10.1038/s41419-023-06193-1.
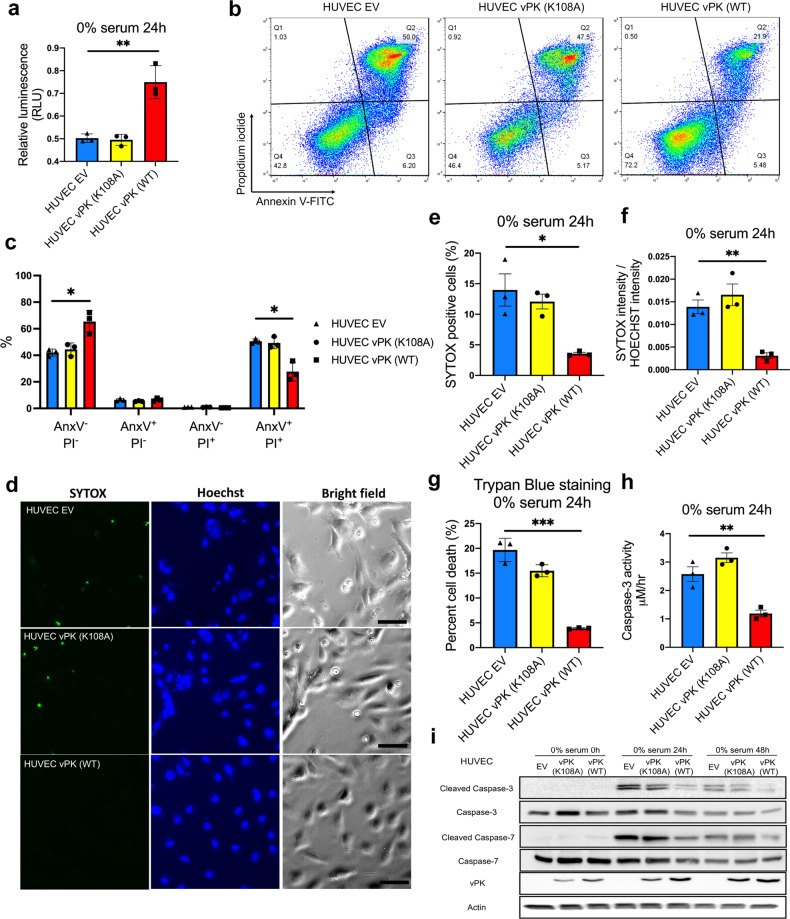
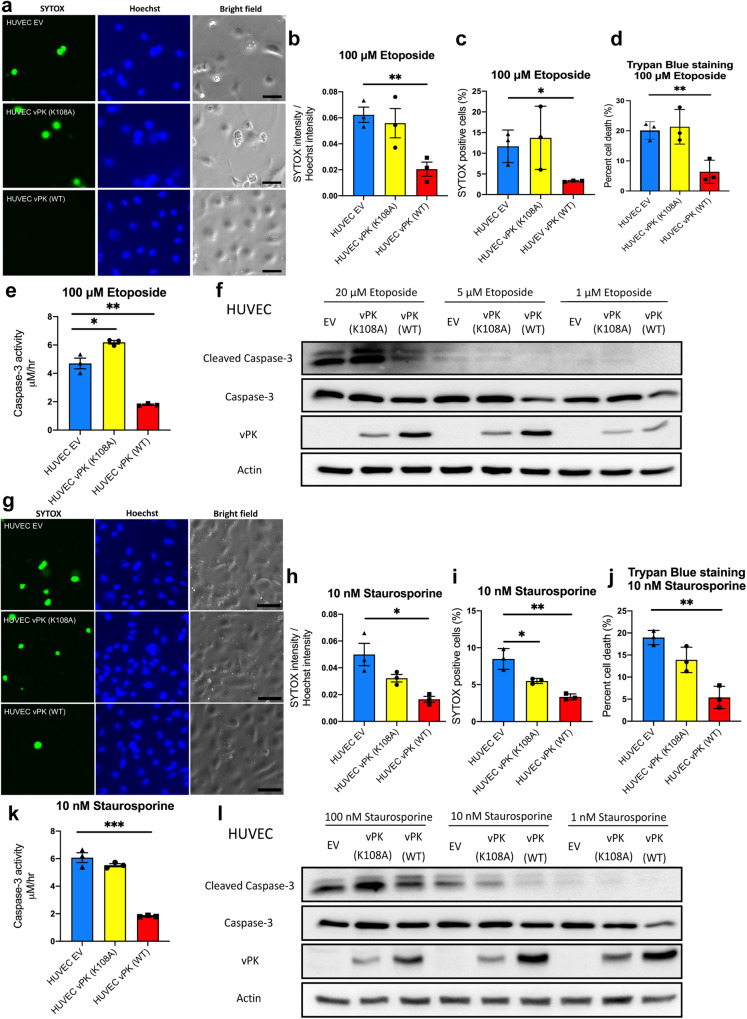
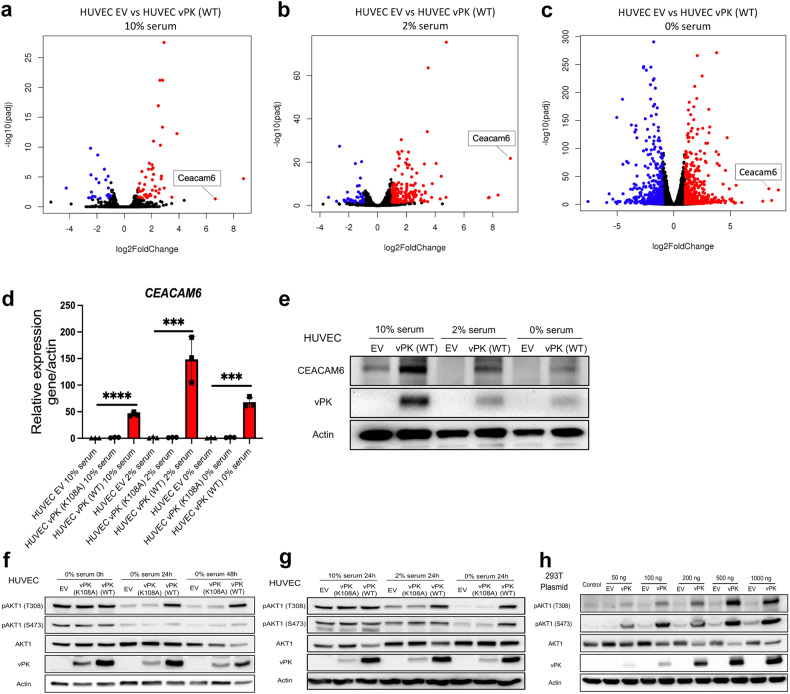
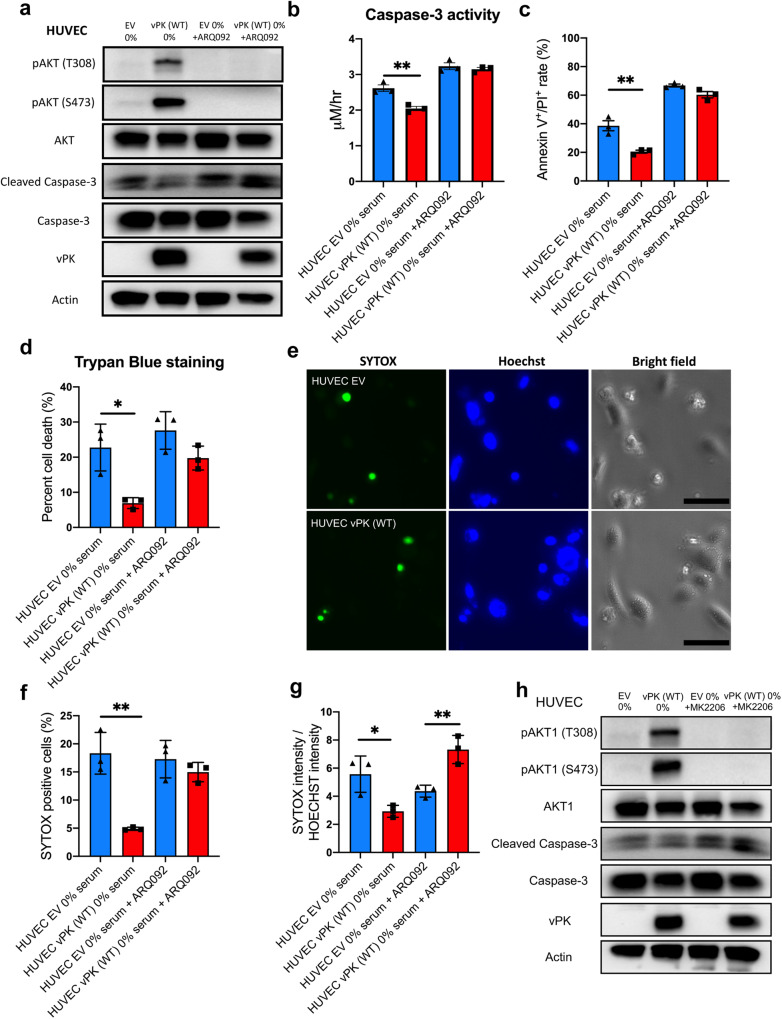
Oncogenic viruses have developed various strategies to antagonize cell death and maintain lifelong persistence in their host, a relationship that may contribute to cancer development. Understanding how viruses inhibit cell death is essential for understanding viral oncogenesis. Kaposi's sarcoma-associated herpesvirus (KSHV) is associated with three different cancers in the human population, including Kaposi's sarcoma (KS), the most common cancer in HIV patients. Previous studies have indicated that the KSHV-encoded viral protein kinase (vPK) impacts many processes dysregulated in tumorigenesis. Here, we report that vPK protects cells from apoptosis mediated by Caspase-3. Human umbilical vein endothelial cells (HUVECs) expressing vPK (HUVEC-vPK) have a survival advantage over control HUVEC under conditions of extrinsic- and intrinsic-mediated apoptosis. Abolishing the catalytic activity of vPK attenuated this survival advantage. We found that KSHV vPK-expressing HUVECs exhibited increased activation of cellular AKT kinase, a cell survival kinase, compared to control cells without vPK. In addition, we report that vPK directly binds the pleckstrin homology (PH) domain of AKT1 but not AKT2 or AKT3. Treatment of HUVEC-vPK cells with a pan-AKT inhibitor Miransertib (ARQ 092) reduced the overall phosphorylation of AKT, resulting in the cleavage of Caspase-3 and the induction of apoptosis. Furthermore, vPK expression activated VEGF/VEGFR2 in HUVECs and promoted angiogenesis through the AKT pathway. vPK expression also inhibited the cytotoxicity of cisplatin in vitro and in vivo. Collectively, our findings demonstrate that vPK's ability to augment cell survival and promote angiogenesis is critically dependent on AKT signaling, which is relevant for future therapies for treating KSHV-associated cancers.
致癌病毒已开发出各种策略来拮抗细胞死亡并在宿主中维持终身持久性,这种关系可能有助于癌症的发展。了解病毒如何抑制细胞死亡对于理解病毒致癌作用至关重要。卡波西肉瘤相关疱疹病毒(KSHV)与人类群体中的三种不同癌症有关,包括卡波西肉瘤(KS),这是 HIV 患者最常见的癌症。先前的研究表明,KSHV 编码的病毒蛋白激酶(vPK)影响肿瘤发生过程中失调的许多过程。在这里,我们报告 vPK 可保护细胞免受 Caspase-3 介导的细胞凋亡。在体外和内在介导的细胞凋亡条件下,表达 vPK 的人脐静脉内皮细胞(HUVEC-vPK)比对照 HUVEC 具有生存优势。消除 vPK 的催化活性会减弱这种生存优势。我们发现,与没有 vPK 的对照细胞相比,表达 KSHV vPK 的 HUVEC 表现出细胞 AKT 激酶(一种细胞存活激酶)的激活增加。此外,我们报告 vPK 可直接结合 AKT1 的 pleckstrin 同源(PH)结构域,但不结合 AKT2 或 AKT3。用 pan-AKT 抑制剂 Miransertib(ARQ 092)处理 HUVEC-vPK 细胞可降低 AKT 的整体磷酸化,导致 Caspase-3 的切割和细胞凋亡的诱导。此外,vPK 表达在 HUVEC 中激活了 VEGF/VEGFR2,并通过 AKT 通路促进血管生成。vPK 表达还抑制了 cisplatin 在体外和体内的细胞毒性。总之,我们的研究结果表明,vPK 增强细胞存活和促进血管生成的能力严重依赖于 AKT 信号,这对于治疗 KSHV 相关癌症的未来疗法具有重要意义。