Department of Genetics, Evolution, and Environment, University College London, London WC1E 6BT, United Kingdom.
Department of Statistics and Data Science, China Southern University of Science and Technology, Shenzhen 518055, China.
Proc Natl Acad Sci U S A. 2023 Oct 31;120(44):e2310708120. doi: 10.1073/pnas.2310708120. Epub 2023 Oct 23.
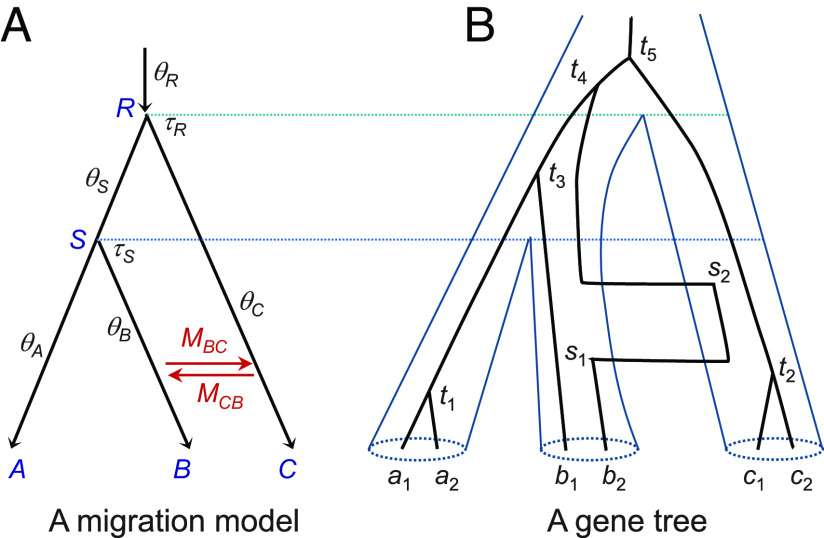
Analyses of genome sequence data have revealed pervasive interspecific gene flow and enriched our understanding of the role of gene flow in speciation and adaptation. Inference of gene flow using genomic data requires powerful statistical methods. Yet current likelihood-based methods involve heavy computation and are feasible for small datasets only. Here, we implement the multispecies-coalescent-with-migration model in the Bayesian program bpp, which can be used to test for gene flow and estimate migration rates, as well as species divergence times and population sizes. We develop Markov chain Monte Carlo algorithms for efficient sampling from the posterior, enabling the analysis of genome-scale datasets with thousands of loci. Implementation of both introgression and migration models in the same program allows us to test whether gene flow occurred continuously over time or in pulses. Analyses of genomic data from mosquitoes demonstrate rich information in typical genomic datasets about the mode and rate of gene flow.
基因组序列数据分析揭示了普遍存在的种间基因流动,并丰富了我们对基因流动在物种形成和适应中的作用的理解。使用基因组数据推断基因流动需要强大的统计方法。然而,目前基于似然的方法涉及大量计算,仅适用于小数据集。在这里,我们在贝叶斯程序 bpp 中实现了多物种合并-迁移模型,该模型可用于检测基因流动和估计迁移率,以及物种分歧时间和种群大小。我们开发了用于从后验中有效抽样的马尔可夫链蒙特卡罗算法,从而能够分析具有数千个基因座的基因组规模数据集。在同一个程序中同时实现基因渗入和迁移模型,使我们能够测试基因流动是持续发生还是脉冲式发生。对蚊子基因组数据的分析表明,典型基因组数据集中包含有关基因流动方式和速率的丰富信息。